
MALDI-TOF Mass Spectrometry for Veterinary Microbial Identification
Overview and Principles of MALDI-TOF Mass Spectrometry for Veterinary Microbial Identification
Fundamental Principles of MALDI-TOF Mass Spectrometry
Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry (MALDI-TOF MS) has fundamentally transformed the landscape of clinical and veterinary microbiology, transitioning from an emerging technology to a cornerstone diagnostic platform over the past two decades [1, 2, 3]. At its core, the technique relies on the generation of characteristic mass spectral fingerprints - primarily derived from highly abundant and conserved ribosomal proteins - to achieve rapid and accurate microbial identification [4, 1, 5]. The underlying principle is elegantly straightforward: microbial cells, either intact or following a simple protein extraction, are co-crystallized with an organic matrix solution on a target plate. Upon irradiation with a pulsed ultraviolet laser, the matrix absorbs the energy, leading to rapid desorption and ionization of the analyte molecules, predominantly ribosomal proteins in the mass range of 2,000 to 20,000 Daltons [6, 1]. These ionized proteins are then accelerated through an electric field and travel down a field-free flight tube under high vacuum. The time-of-flight (TOF) of each ion is inversely proportional to the square root of its mass-to-charge ratio (m/z), with smaller ions reaching the detector more rapidly than larger ones. The resulting spectrum, a plot of ion intensity versus m/z, constitutes a unique and reproducible molecular fingerprint for a given microorganism [1, 5].
The power of MALDI-TOF MS for microbial identification lies not in the sequencing of individual proteins but in the pattern recognition of the entire spectral profile. This profile is compared against a curated reference database of Main Spectra Profiles (MSPs) generated from well-characterized, often type-strain, microorganisms [7, 8, 1]. Sophisticated algorithms calculate a logarithmic identification score based on the degree of spectral matching, providing a high-confidence identification at the genus and, frequently, the species level [7, 9]. The entire process, from a single colony on an agar plate to a definitive identification, can be accomplished in under 20 minutes, representing a paradigm shift from traditional phenotypic methods that require 24 to 48 hours or more [4, 9, 10]. This dramatic reduction in turnaround time has profound implications for patient management, antimicrobial stewardship, and outbreak response in both human and veterinary medicine [10, 11].
The Biological Basis: Ribosomal Proteins as Ideal Biomarkers
The exceptional success of MALDI-TOF MS for microbial identification is predicated on the selection of its target analytes. The technique predominantly captures signals from ribosomal proteins, which are ideally suited for this purpose for several compelling biological reasons [4, 1, 5]. Ribosomes are essential, universally conserved organelles responsible for protein synthesis, and their constituent proteins are expressed at high levels in all actively growing cells. This high abundance ensures robust and reproducible signals, even from a relatively small number of bacterial cells. Ribosomal proteins are under strong evolutionary constraints, meaning their sequences are highly conserved within a species yet exhibit sufficient inter-species variability to serve as reliable taxonomic markers [1]. This provides the necessary discriminatory power to differentiate between closely related species, a task that can be challenging for traditional biochemical methods. The mass range of these proteins (typically 4,000-15,000 Da) is also ideal for standard MALDI-TOF MS instruments, allowing for high-resolution spectral acquisition without the need for complex sample preparation or protein digestion [6, 1].
While the protein-based approach is the gold standard, the detection of other molecular classes, such as lipids, is an area of active research and expanding capability. Lipids, including phospholipids and lipopolysaccharides, are also abundant and structurally diverse components of the microbial cell envelope. Their analysis by MALDI-TOF MS can provide complementary information, particularly for organisms where protein-based identification is challenging, such as mycobacteria or for discriminating between closely related species like Escherichia coli and Shigella spp. [12]. Lipid profiles can be directly linked to antimicrobial resistance mechanisms, such as the modification of lipid A with phosphoethanolamine or aminoarabinose in polymyxin-resistant Gram-negative bacteria, offering a direct readout of a resistance phenotype [12]. This dual capability - identification and resistance marker detection from the same analytical platform - highlights the evolving sophistication of MALDI-TOF MS.
The Analytical Workflow: From Sample to Identification
The standard workflow for MALDI-TOF MS-based microbial identification in a veterinary diagnostic laboratory is characterized by its simplicity and speed, but it is not without critical nuances that demand technical rigor [13]. The process begins with the acquisition of a pure microbial isolate, typically a single colony from a primary culture plate. The most common and rapid method is the "direct transfer" or "whole-cell" method, where a small amount of the colony is smeared directly onto a target plate and overlaid with a saturated solution of an appropriate matrix, most commonly α-cyano-4-hydroxycinnamic acid (HCCA) in a mixture of organic solvent, water, and trifluoroacetic acid [1, 5]. The matrix solution simultaneously extracts proteins from the cells and facilitates their co-crystallization upon drying. For more fastidious organisms, such as Gram-positive bacteria with thick cell walls, yeasts, or mycobacteria, an additional "on-plate" or "tube-based" extraction step using formic acid and acetonitrile is often necessary to achieve efficient protein extraction and higher identification scores [14, 11]. For highly pathogenic bacteria (HPB), such as Bacillus anthracis or Francisella tularensis, specialized inactivation protocols that are compatible with MALDI-TOF MS analysis are critical for laboratory safety [8].
Once the sample is prepared, the target plate is introduced into the mass spectrometer. The instrument automatically acquires spectra from multiple positions on each sample spot, accumulating hundreds of laser shots to generate a final, averaged spectrum. This spectrum is then processed by the instrument's software - subtracting baseline noise, smoothing, and peak picking - before being compared against the reference database [13, 1]. The result is a numerical score that indicates the confidence of the identification. For the widely used Bruker Biotyper system, a score of ≥2.30 indicates highly probable species-level identification, a score of 2.00-2.29 indicates secure genus identification with probable species identification, a score of 1.70-1.99 indicates probable genus identification, and a score of <1.70 is considered unreliable [7, 15]. The VITEK MS system (bioMérieux) uses a different confidence value, but the principle of spectral matching against a curated database is identical [16]. This standardized scoring system provides an objective and reproducible metric for identification, a significant advantage over the subjective interpretation of many traditional tests.
Database Dependency and the Imperative for Expansion
The accuracy and scope of MALDI-TOF MS are inextricably linked to the quality and comprehensiveness of the reference spectral database used for comparison [1, 5]. Commercial databases, such as those provided by Bruker Daltonics and bioMérieux, are extensive and cover the vast majority of clinically relevant human pathogens. However, their coverage of veterinary-specific, environmental, and emerging pathogens is often less complete [17, 13, 18]. This limitation is a critical consideration for veterinary microbiology, where the diversity of potential pathogens is immense, spanning companion animals, livestock, poultry, aquatic species, and wildlife. A failure to identify an isolate, or a misidentification, can have serious consequences for animal health, food safety, and public health.
Numerous studies have demonstrated that the addition of custom, in-house generated MSPs to the commercial database can dramatically improve identification rates for veterinary-relevant organisms. For instance, the creation of a custom database for Aeromonas spp. isolated from diseased fish increased the rate of accurate species-level identification from a mere 5.3% using the standard library to 100% [7]. Similarly, the identification of zoophilic dermatophytes, such as Trichophyton erinacei from hedgehogs, was only possible after the development of an in-house database supplemented with spectra from these specific isolates [18]. The identification of non-tuberculous mycobacteria (NTM) from various animal species also benefits significantly from expanded databases, as commercial libraries may lack spectra for many of the over 200 NTM species [14]. For fungal identification, a dedicated web application and an in-house database of reference spectra proved far superior to conventional morphological methods, correctly identifying 89% of veterinary fungal isolates compared to only 60% by traditional approaches [17]. These examples underscore a fundamental principle: the successful implementation of MALDI-TOF MS in a veterinary setting is not a "plug-and-play" endeavor but requires a proactive commitment to database curation and expansion to reflect the unique microbial ecology of the target animal populations [13]. Collaborative initiatives, such as the publicly available database for highly pathogenic bacteria developed by Lasch et al. [8], and platforms like the ROVETEMERG system [13], which integrates Bruker libraries with public health databases like CDC MicrobeNet, represent important steps toward overcoming this limitation.
Addressing the Challenges: Closely Related Species, Misidentification, and Emerging Pathogens
Despite its transformative impact, MALDI-TOF MS is not without its challenges. The most significant limitation is the difficulty in discriminating between very closely related species that share highly similar ribosomal protein profiles [19, 6]. This is a well-recognized issue for several clinically important groups. For example, the technique struggles to reliably differentiate Escherichia coli from Shigella species, as they are essentially the same species from a genomic perspective [12]. Similarly, members of the Streptococcus mitis group, Enterobacter cloacae complex, and certain Corynebacterium species can be difficult to resolve at the species level using standard MALDI-TOF MS [20, 21, 22]. In the veterinary context, misidentification of Raoultella spp. and Klebsiella variicola as Klebsiella pneumoniae has been documented in equine necropsy samples, highlighting the potential for error when relying solely on commercial databases [23]. The initial misidentification of Bordetella pseudohinzii from wild rodents as B. avium or B. hinzii by MALDI-TOF MS further illustrates this point, with whole-genome sequencing ultimately required for definitive species assignment [24].
To overcome these challenges, several advanced strategies are being developed. One approach involves the use of machine learning (ML) and deep learning algorithms to analyze the full spectral information, rather than just the peak lists used by conventional software [25, 26, 27]. These algorithms can identify subtle, sub-phenotypic differences in the spectra that are invisible to the human eye or standard pattern-matching algorithms. For instance, an XGBoost model was successfully developed to differentiate eight Salmonella serotypes, a task that is notoriously difficult for standard MALDI-TOF MS [25]. A convolutional neural network (CNN) was able to identify a clonal population of Aspergillus flavus with high accuracy, demonstrating the potential for strain-level typing and outbreak detection [26]. Another promising avenue is the use of "peptide mass fingerprinting" following a targeted tryptic digest of extracted proteins. This approach, often coupled with liquid chromatography (LC-MALDI-TOF/TOF), extends the analysis to a larger number of peptides, providing greater discriminatory power for subspecies-level identification [4]. The detection of species-specific lipids, as previously mentioned, offers another orthogonal layer of information that can resolve ambiguities [12]. These technological advancements are rapidly expanding the capabilities of MALDI-TOF MS, pushing its utility beyond simple species identification toward more sophisticated applications like strain typing and antimicrobial resistance profiling.
Applications in Veterinary Microbiology: A One Health Perspective
The application of MALDI-TOF MS in veterinary microbiology is broad and continues to expand, driven by the need for rapid, accurate, and cost-effective diagnostics. Its utility has been demonstrated across many animal species and sample types. In food animal production, it is used for the identification of mastitis pathogens in dairy cattle, enabling rapid and targeted therapy [28, 29, 15]. The technique has been instrumental in identifying emerging mastitis-associated pathogens like Corynebacterium parakroppenstedtii and C. pseudokroppenstedtii [20]. In aquaculture, MALDI-TOF MS is a powerful tool for the rapid identification of bacterial pathogens such as Aeromonas spp., Streptococcus spp., and Vibrio spp., which cause significant economic losses [7, 30]. Its speed is critical for monitoring and controlling outbreaks of notifiable diseases, such as those caused by Viral Hemorrhagic Septicemia Virus or Infectious Hematopoietic Necrosis Virus, where rapid differential diagnosis from bacterial infections is paramount. In companion animal medicine, it aids in the diagnosis of urinary tract infections, skin and ear infections, and periodontal disease [31, 32, 33]. The technique is also proving valuable in wildlife health surveillance, enabling the identification of pathogens in a diverse range of species, from marine polychaetes to wild rodents and hybrid marmosets [34, 35, 24].
Crucially, the role of MALDI-TOF MS extends beyond individual animal diagnosis to encompass the broader One Health framework. The technique is a cornerstone for antimicrobial resistance (AMR) surveillance, as it can rapidly identify bacterial species that are known reservoirs of resistance genes [36, 37, 30]. By providing rapid species identification, it facilitates the early detection of zoonotic pathogens in animal populations, food products, and the environment, thereby informing public health risk assessments [38, 39, 40]. The ability to quickly and accurately identify pathogens like Avian Influenza Virus, West Nile Virus in Birds, or Rabies Lyssavirus is critical for outbreak control, but for bacterial pathogens, MALDI-TOF MS is the frontline tool. The integration of MALDI-TOF MS data with genomic and epidemiological information, as advocated by organizations like the World Organisation for Animal Health (WOAH), strengthens global surveillance networks and supports the implementation of effective control strategies [36, 41]. The technology's high throughput and low per-sample cost make it ideally suited for large-scale screening programs, a key component of a proactive One Health approach to infectious disease management.
Instrumentation and Methodological Workflow for Veterinary Sample Analysis and Extraction
The successful deployment of Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry (MALDI-TOF MS) in veterinary clinical microbiology rests upon a meticulously optimized instrumentation platform and a standardized, reproducible methodological workflow. As a leading veterinary clinical pathologist, I must emphasize that the fidelity of species-level identification is inextricably linked to the rigor applied at each pre-analytical, analytical, and post-analytical step. This section provides an exhaustive, mechanism-level examination of the hardware, sample preparation protocols, and data acquisition strategies that constitute the operational backbone of veterinary MALDI-TOF MS, drawing upon a wealth of recent empirical evidence to guide best practices.
Instrumentation Core: The Mass Spectrometer and Laser System
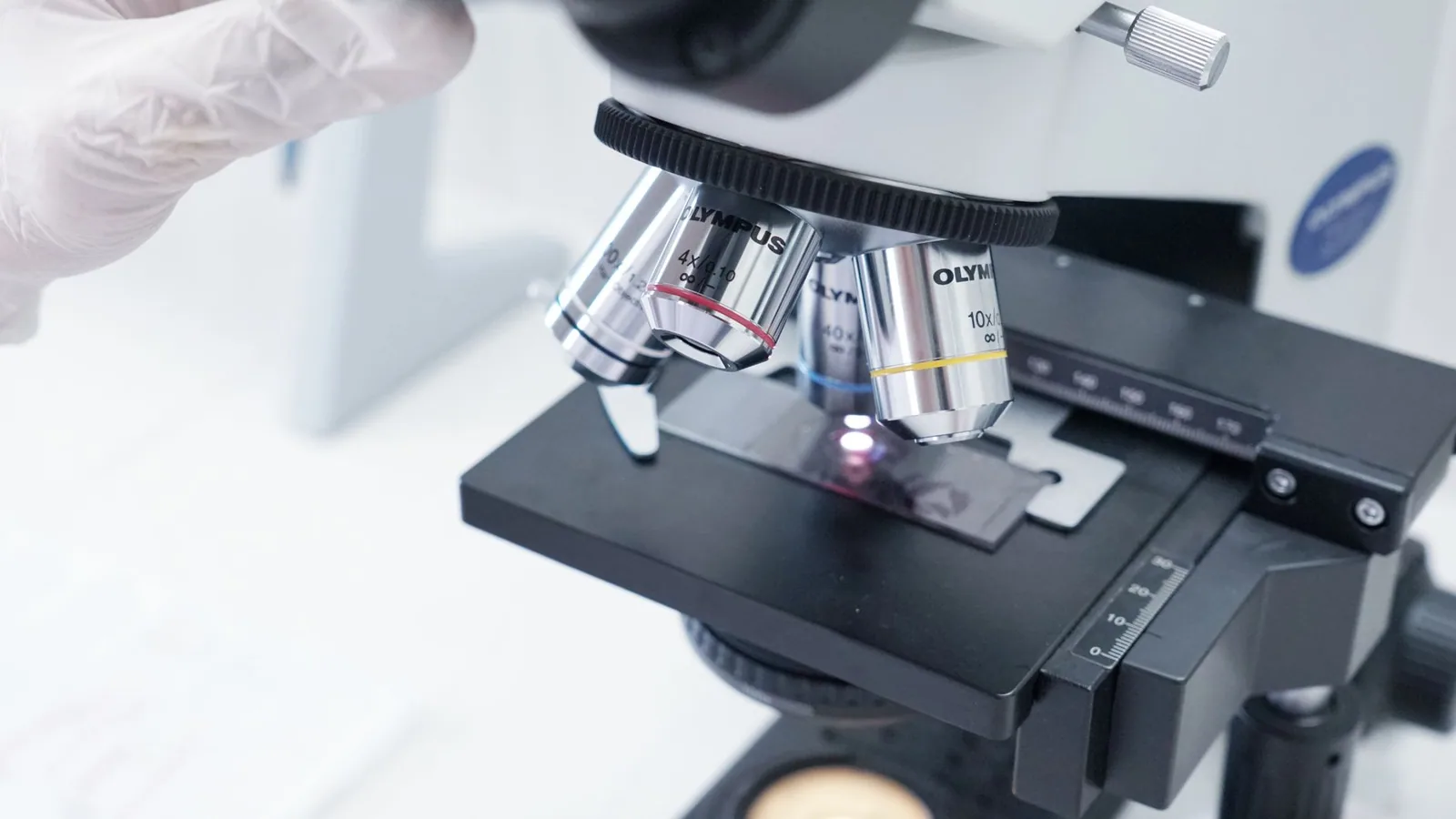
The foundational hardware for veterinary MALDI-TOF MS is the time-of-flight mass spectrometer, most commonly represented by commercial platforms such as the Bruker MALDI Biotyper (Sirius One, Microflex series) and the bioMérieux Vitek MS systems [13, 6, 2]. The core principle involves the pulsed irradiation of a co-crystallized sample-matrix mixture on a polished steel target plate with a nitrogen laser (typically 337 nm) or a solid-state laser (355 nm). The laser energy is absorbed primarily by the matrix - most often α-cyano-4-hydroxycinnamic acid (HCCA) for microbial protein profiling - which undergoes rapid desorption and ionization, transferring protons to the analytes [1]. This process yields predominantly singly charged ions ([M+H]⁺) of abundant, low-molecular-weight proteins, particularly ribosomal proteins (ca. 2,000-20,000 Da), which serve as the taxonomic fingerprints [6, 1]. The ions are then accelerated in an electric field and travel through a field-free flight tube under high vacuum. The time-of-flight (TOF) is inversely proportional to the square root of the mass-to-charge ratio (m/z), allowing the detector to generate a mass spectrum comprising hundreds of peaks, the pattern of which is characteristic of a given microbial species [1, 3].
Modern instruments have evolved significantly. The Bruker MALDI Biotyper Sirius One, now operational at specialized veterinary platforms like ROVETEMERG, features enhanced sensitivity and mass accuracy through improved ion optics and a faster laser repetition rate, enabling the acquisition of high-quality spectra from as few as 10⁴-10⁵ colony-forming units [13]. Critically, these systems are calibrated daily using a bacterial test standard (BTS; e.g., Escherichia coli DH5α) to ensure mass accuracy within 200-400 ppm, a prerequisite for reliable database matching [13, 5]. The integration of automated target plate loaders and batch-processing software has dramatically increased throughput, allowing a single technician to process over 100 isolates per hour - a paradigm shift from the 24-48 h required for conventional biochemical panels [6, 10]. This speed is particularly transformative for veterinary applications, where rapid etiological diagnosis is critical for outbreak containment in livestock herds, aquaculture facilities, and companion animal clinics [13, 10].
Sample Preparation Protocols: From Colony to Crystalline Matrix
The quality of the MALDI-TOF MS spectrum is fundamentally determined by the sample preparation method. For routine veterinary isolates, a direct smear or "direct transfer" (DT) protocol is the fastest approach, involving the application of a small amount of fresh bacterial colony (<24 h growth) directly onto the target plate, overlaying it with 1 μL of 70% formic acid for on-plate protein extraction (especially for Gram-positive bacteria), and then allowing it to dry before adding 1 μL of matrix solution (saturated HCCA in 50% acetonitrile/2.5% trifluoroacetic acid) [13, 5, 3]. For most Enterobacteriaceae, Staphylococcaceae, and Enterococcaceae, this rapid method yields log scores ≥2.0, signifying high-confidence species identification [7, 13, 6, 15]. However, the DT protocol may prove insufficient for organisms with robust cell walls, such as yeasts, filamentous fungi, and mycobacteria, or for isolates that produce significant extracellular polysaccharide [14, 17, 6].
For these recalcitrant taxa, a full "tube-based" extraction is mandatory. The standardized protocol involves mechanical disruption of cells (e.g., bead-beating for mycobacteria) or chemical lysis with ethanol, followed by sequential treatment with 70% formic acid and 100% acetonitrile [14, 1, 3]. After centrifugation, the protein-rich supernatant is spotted onto the target plate, dried, and overlaid with matrix. This extraction procedure efficiently liberates ribosomal proteins from intracellular compartments while removing salts and other low-molecular-weight contaminants that can suppress ionization. In a multicenter evaluation of highly pathogenic bacteria, Lasch et al. [8] demonstrated that a standardized inactivation/extraction protocol (using 70% ethanol and 70% formic acid) yielded high-quality spectra for 1,601 strains across 264 species, including Category A agents, validating the universal applicability of the approach for biosafety Level 3 organisms. For veterinary mycobacteria, Lorente-Leal et al. [14] reported that a modified bead-beating extraction (using 0.5 mm zirconia beads) prior to formic acid/acetonitrile extraction achieved an identification rate of 76% for non-tuberculous mycobacteria from diverse animal hosts, with spectra being highly reproducible across replicates.
Specialized Workflows for Complex Veterinary Matrices
Veterinary samples pose unique challenges compared to human clinical specimens. The presence of mixed flora, high lipid content (e.g., from milk in mastitis diagnostics), and antimicrobial residues can compromise spectral quality. For bovine mastitis, Santos et al. [15] optimized a workflow involving initial culture on 5% sheep blood agar for 18-24 h, followed by the formic acid extraction method. They achieved a species-level identification rate of 99.4% for 321 isolates, including uncommon species like Burkholderia cepacia and Ralstonia pickettii [15]. Critically, the use of a short incubation time is essential; extended culture (>48 h) can lead to sporulation and altered protein expression, reducing spectral matching scores [6, 15].
For aquatic animal pathogens, such as Aeromonas spp. from diseased fish, Mursalim et al. [7] developed a custom workflow that involved direct colony spotting followed by a 70% formic acid overlay. This method, when paired with a species-specific main spectra profile (MSP) database, increased the genus-level identification rate to 100% and species-level to 100% for Aeromonas isolates, compared to only 5.3% using the standard commercial database [7]. This underscores the critical need for veterinary-specific spectral libraries, particularly for under-represented aquatic taxa like Channel Catfish Virus (though the bacterial pathogens, not the virus itself, are identified via this technique), and for pathogens like Streptococcus agalactiae from buffalo or cattle mastitis [29]. Pereira et al. [29] demonstrated that MALDI-TOF MS could successfully group S. agalactiae isolates from subclinical mastitis into 8 distinct clusters, correlating with antimicrobial susceptibility profiles, thereby providing epidemiological insights beyond simple identification.
Direct-from-Sample Workflows: Blood Cultures and Urine
A major advancement in veterinary microbiology is the application of MALDI-TOF MS directly from positive blood culture broth, bypassing the need for subculture and saving 12-24 h. Broyer et al. [16] evaluated an automated sample preparation instrument (Vitek MS prototype) that uses a specialized extraction strip containing reagents to lyse blood cells and extract bacterial proteins directly from BacT/ALERT bottles. The instrument achieved 87% species-level identification for Gram-negative bacteria, 88% for Gram-positive bacteria, and 100% for yeasts, using the Vitek MS database [16]. In a veterinary context, this approach is highly translatable to equine, canine, and feline sepsis cases. For direct blood testing, Cortazzo et al. [42] reported the first pediatric case of bacterial identification directly from whole blood using MALDI-TOF MS, although this remains experimental for routine veterinary use.
For urine samples, bacterial concentration steps (e.g., centrifugation at 10,000 × g for 10 min, followed by washing with distilled water and formic acid extraction) can yield reliable spectra. Moreno et al. [32] successfully identified Escherichia coli and other uropathogens from sow urine using this method, revealing a high prevalence of multidrug-resistant strains. Similarly, for ocular samples in microbial keratitis, Kato et al. [43] demonstrated that pediatric blood culture bottles, when combined with MALDI-TOF MS identification of the recovered isolates, achieved a sensitivity of 41.3% compared to 43.3% for conventional multi-sampling, with substantial diagnostic agreement (kappa = 0.69). This protocol is particularly advantageous when sample volume is limited.
Database Construction and Spectral Acquisition Parameters
The performance of MALDI-TOF MS is entirely dependent on the quality and breadth of the reference spectral library. Commercial databases (Bruker BDAL, Vitek MS IVD) are heavily weighted toward human clinical pathogens, leaving significant gaps for veterinary-specific, environmental, and aquatic pathogens [21, 23, 17, 13, 18]. Gravey et al. [23] highlighted that misidentification of Raoultella terrigena and Klebsiella grimontii as Klebsiella pneumoniae occurred because these species lacked adequate reference spectra in the commercial library. Similarly, Zhou et al. [24] found that initial MALDI-TOF MS misclassified Bordetella pseudohinzii from wild rodents as B. avium or B. hinzii, with correct identification only achieved after whole-genome sequencing.
To address this, a rigorous protocol for in-house MSP construction has been established. Each MSP is typically built from 20-40 high-quality spectra acquired from at least three independent cultures of a well-characterized reference strain (identified by 16S rRNA or whole-genome sequencing) [7, 8]. Spectra are acquired in linear positive ion mode across an m/z range of 2,000-20,000 Da. Specific acquisition parameters include: laser frequency of 60 Hz, ion source 1 voltage of 20 kV, ion source 2 voltage of 18.0 kV, lens voltage of 7.5 kV, and a detector gain that is optimized to avoid saturation [8, 13]. Each spectrum is the sum of 240-500 laser shots at varying positions within the target spot to account for heterogeneity in crystal morphology. Following baseline subtraction and smoothing, the spectra are processed using a peak detection algorithm (e.g., the MALDI Biotyper Compass Explorer), and the resulting peak list (intensity and m/z) is used to generate an MSP containing the most reliable and reproducible peaks [8, 13].
The Rubini Laboratory (publicly available via ZENODO) has made significant contributions by releasing a database containing 11,055 spectra from 1,601 strains of 264 species, with a focus on highly pathogenic bacteria [8]. This resource is invaluable for veterinary laboratories dealing with zoonotic agents like Brucella canis (though not in the provided list, the general principle applies) or Bacillus anthracis. For fungal identification, Becker et al. [17] validated an online web application containing reference spectra for 69 fungal species from animals, achieving 89% correct species-level identification compared to only 60% using conventional methods. The construction of fungal MSPs requires additional steps: homogenization of mycelial mats in 70% ethanol, mechanical lysis using a bead mill, and extraction with formic acid/acetonitrile [17, 26].
Bioinformatics and Data Interpretation: From Raw Spectra to Clinical Action
The final, and perhaps most intellectually demanding, stage is the bioinformatic interpretation of the acquired spectra. The MALDI Biotyper algorithm (also used by Vitek MS with a modified commercial database) compares the unknown spectrum to each reference MSP in the library using a pattern-matching algorithm based on the number of matched peaks, peak intensities, and mass tolerances. The output is a log(score) value ranging from 0 to 3.0. The standard interpretive criteria are: log(score) ≥ 2.0 for high-confidence species identification; ≥ 1.7 but < 2.0 for probable genus identification; and < 1.7 for no reliable identification [13, 6, 15]. However, these thresholds are empirically derived for human clinical databases. For veterinary applications, particularly with custom databases, lower cutoffs may be acceptable. Mursalim et al. [7] used a cutoff of ≥ 2.3 with their custom Aeromonas database to achieve 100% species-level confidence, while the standard 2.0 threshold still yielded 100% genus-level identification.
Beyond simple matching, advanced data analysis is critical for strain typing, detection of antimicrobial resistance (AMR), and cluster analysis. Dendrograms can be constructed based on the similarity of MSPs using correlation-based distance metrics (e.g., Bruker's MS-based clustering tool). Pereira et al. [29] demonstrated that MALDI-TOF MS grouping of S. agalactiae isolates from mastitis produced 8 clusters that correlated with herd of origin and - importantly - with antimicrobial susceptibility profiles, identifying cluster-specific resistance to enrofloxacin and cephalexin. This capability has direct One Health implications: a veterinary laboratory can detect a clonal spread of a multidrug-resistant Staphylococcus aureus strain from cows with subclinical mastitis within hours, enabling immediate implementation of enhanced biosecurity measures.
Principal Component Analysis (PCA) and machine learning (ML) algorithms are increasingly applied to MALDI-TOF MS data to discriminate between closely related species and to predict AMR. Sipahi and Çevik [44] used PCA to differentiate extended-spectrum β-lactamase (ESBL)-producing E. coli from susceptible E. coli based on subtle differences in their protein profiles, with the ESBL-producing strains clustering separately. Ren et al. [25] developed an XGBoost model using 16 MALDI-TOF MS spectral features that achieved an AUC of 0.9898 for serotyping Salmonella (including S. Typhimurium and S. Enteritidis), a task previously impossible with conventional MALDI-TOF MS databases. The integration of deep learning convolutional neural networks (CNNs) has also been reported for the detection of clonal Aspergillus flavus strains [26] and for improved discrimination of closely related microbial species [19]. These ML approaches are transitioning from research to routine use, with the caveat that model validation on external datasets remains a challenge [27].
The instrumentation and methodological workflow for veterinary MALDI-TOF MS represents a multi-layered system where instrument calibration, tailored sample preparation, species-specific database enrichment, and bioinformatic interpretative algorithms converge. The clinical pathologist must understand the biological rationale behind each step - from the laser-facilitated desorption of ribosomal proteins to the pattern-recognition algorithms that decode them - to troubleshoot suboptimal identifications and to confidently report results that guide antimicrobial therapy and infection control across the One Health continuum. The ongoing expansion of veterinary-specific spectral libraries, coupled with AI-driven analytical pipelines, promises to further elevate the diagnostic precision of this transformative technology for pathogens as diverse as Avian Influenza Virus and Porcine Epidemic Diarrhea Virus [13, 41].
Spectral Data Analysis and Bioinformatics Algorithms for Microbial Classification and Identification
The transformation of raw mass spectral data into a definitive taxonomic assignment represents the computational cornerstone of MALDI-TOF MS-based microbial identification. This process, far from being a trivial pattern-matching exercise, involves a sophisticated pipeline of signal processing, feature extraction, and algorithmic classification that determines the accuracy, reliability, and discriminatory power of the entire diagnostic system. In the veterinary context, where pathogen diversity spans across domesticated livestock, companion animals, wildlife, and aquatic species, the robustness of these bioinformatics algorithms becomes paramount for ensuring that clinically actionable identifications are delivered with confidence.
Spectral Preprocessing and Peak Detection Algorithms
The journey from a raw mass spectrum to a usable microbial fingerprint begins with extensive preprocessing. The raw spectrum, acquired across a mass-to-charge (m/z) range typically spanning 2,000 to 20,000 Da for ribosomal protein profiling, is contaminated with baseline drift, electronic noise, and matrix-related signals that must be systematically removed [1, 5]. Sophisticated algorithms first perform baseline correction, often employing a rolling-disk or spline-fitting approach that estimates and subtracts the continuous background signal. Smoothing algorithms, such as the Savitzky-Golay filter, are then applied to reduce high-frequency noise while preserving the integrity of true ion signals. Normalization procedures adjust for inter-spectrum variability in total ion current, ensuring that spectra acquired from different cultures, different days, or even different instruments can be meaningfully compared [19, 6].
Peak detection represents the critical juncture where continuous spectral data is converted into a discrete list of m/z values and their corresponding intensities. Algorithms must distinguish true biological signals from noise, a challenge that becomes particularly acute for low-abundance proteins or for spectra acquired from suboptimal culture conditions. Most commercial platforms, including the Bruker MALDI Biotyper and the bioMérieux VITEK MS, employ proprietary peak detection algorithms that apply signal-to-noise ratio thresholds, typically requiring peaks to exceed a factor of three to six times the local noise level [6, 11]. The detected peaks are then subjected to mass calibration correction using internal or external standards, ensuring that m/z values are accurate to within approximately 200-400 parts per million. This level of precision is essential because the ribosomal proteins that form the basis of identification have highly conserved molecular weights across strains of the same species, and even small mass shifts can lead to misclassification [5, 3].
Pattern Matching and Database Comparison Algorithms
The core identification algorithm in commercial MALDI-TOF MS systems operates on the principle of pattern matching against a curated reference database of main spectra profiles (MSPs). Each MSP is a composite spectrum generated from multiple replicate measurements of a well-characterized reference strain, containing the list of most representative peaks with their average masses and relative intensities [7, 6]. The Bruker MALDI Biotyper system, for instance, employs a proprietary algorithm that calculates a log(score) value ranging from 0 to 3.0, reflecting the degree of similarity between the unknown spectrum and each reference MSP in the database. A log(score) of ≥2.0 indicates secure genus identification, while ≥2.3 indicates highly probable species identification [7, 31, 15]. These thresholds, however, are not absolute; they were empirically derived from large validation studies and may require adjustment for specific taxonomic groups or for veterinary applications where database coverage may be less comprehensive.
The mathematical underpinnings of these matching algorithms vary between platforms. Some employ correlation-based similarity measures, comparing the entire spectral profile including peak intensities, while others use binary peak lists that consider only the presence or absence of specific m/z values [19, 6]. The VITEK MS system from bioMérieux uses a different algorithmic approach, employing a supervised classification engine known as the Advanced Spectra Classifier, which incorporates both peak presence and intensity information into a multi-dimensional similarity space [16, 6]. Regardless of the specific implementation, all commercial algorithms face the fundamental challenge of distinguishing between closely related species that share highly similar ribosomal protein profiles. This is particularly problematic for members of species complexes, such as those within the Burkholderia cepacia complex, the Mycobacterium tuberculosis complex, or the Streptococcus mitis group, where traditional MALDI-TOF MS algorithms frequently fail to achieve species-level resolution [14, 12, 11].
The Critical Role of Database Curation and Expansion
The accuracy of any pattern-matching algorithm is fundamentally limited by the quality and comprehensiveness of the reference database against which unknown spectra are compared. Commercial databases, while extensive for human clinical pathogens, often lack representation of veterinary-specific organisms, leading to identification failures or, worse, misidentifications [7, 20, 14, 17]. The consequences of inadequate database coverage are well-documented in the veterinary literature. For example, a study on Aeromonas species isolated from diseased fish found that the standard Bruker Biotyper library achieved only 5.3% species-level identification, whereas the incorporation of custom MSPs from reference strains improved species-level identification to 100% [7]. Similarly, the identification of Corynebacterium parakroppenstedtii and C. pseudokroppenstedtii from bovine mastitis cases required the integration of whole-genome sequencing data to resolve species-level classifications that MALDI-TOF MS alone could not achieve [20].
The challenge is particularly acute for non-tuberculous mycobacteria (NTM), where the complex cell wall composition necessitates specialized extraction protocols and where database coverage remains sparse for veterinary isolates. A study evaluating MALDI-TOF MS for NTM identification in veterinary samples reported that only 76% of isolates were identified with high confidence, with 17.3% requiring Sanger sequencing for resolution to the complex level [14]. For zoophilic dermatophytes, such as Trichophyton erinacei and T. verrucosum, commercial databases frequently yield scores only around the cutoff value for secure species identification, necessitating the construction of in-house reference spectra from locally circulating strains [18]. These findings underscore a fundamental principle: the bioinformatics algorithms are only as good as the data they are trained on, and veterinary diagnostic laboratories must invest in database expansion as an ongoing quality improvement activity.
Machine Learning and Deep Learning Approaches
The limitations of traditional pattern-matching algorithms have catalyzed the development of machine learning (ML) and deep learning (DL) approaches that can extract more discriminatory information from MALDI-TOF mass spectra [25, 26, 41, 27]. These methods move beyond simple peak matching to consider the entire spectral profile, including peak shapes, relative intensities, and correlations between peaks, potentially revealing subtle taxonomic differences that are invisible to conventional algorithms. A systematic review of ML applications for MALDI-TOF MS analysis identified support vector machines (SVM), genetic algorithms, artificial neural networks, and random forests as the most frequently employed classifiers, with applications spanning both species identification and antimicrobial resistance prediction [27].
One of the most promising applications of ML in veterinary MALDI-TOF MS is the differentiation of Salmonella serotypes. Traditional serotyping by agglutination is time-consuming, technically demanding, and often inconclusive. A recent study employing an XGBoost algorithm on MALDI-TOF spectra from 692 Salmonella isolates achieved an area under the receiver operating characteristic curve (AUC) of 0.9898 for the training set and 0.9778 for external validation, enabling accurate discrimination of eight clinically relevant serotypes including Salmonella Typhimurium and Salmonella Enteritidis [25]. The study employed a feature selection pipeline that reduced 192 initial spectral features to 16 discriminatory peaks, demonstrating that ML can identify biologically relevant biomarkers that may correspond to serotype-specific proteins or post-translational modifications.
Deep learning, particularly convolutional neural networks (CNNs), has shown remarkable capability for detecting clonal populations within a species. In a study of Aspergillus flavus isolates, a simple CNN trained on MALDI-TOF spectra achieved >93% accuracy in distinguishing a clonal outbreak strain from genetically diverse isolates of the same species, even when spectra were acquired on different instruments [26]. This capability has profound implications for veterinary outbreak investigations, where the ability to rapidly identify clonal transmission of pathogens such as Avian Influenza Virus or Newcastle Disease Virus could inform timely intervention strategies. However, the study also revealed a critical limitation: the CNN's accuracy dropped to 69% when applied to spectra from an older instrument with a degraded laser, highlighting the sensitivity of DL models to variations in spectral quality and the need for rigorous instrument standardization [26].
Algorithms for Antimicrobial Resistance Detection
Beyond species identification, bioinformatics algorithms are being developed to predict antimicrobial resistance (AMR) phenotypes directly from MALDI-TOF spectra, a capability that could revolutionize veterinary antimicrobial stewardship. The biological rationale is that resistance mechanisms, whether mediated by enzymatic degradation, target modification, or efflux pumps, may produce detectable changes in the proteomic or lipidomic profile of the organism [12, 27, 44, 45]. For example, the detection of lipid A modifications, such as the addition of phosphoethanolamine or aminoarabinose residues, can be used as a readout for polymyxin resistance in Gram-negative pathogens [12]. Similarly, the presence of β-lactamase enzymes may be inferred from spectral peaks corresponding to the enzyme's molecular weight, although the low abundance of these proteins relative to ribosomal proteins makes direct detection challenging [44, 45].
Machine learning approaches for AMR prediction typically involve training classifiers on spectra from phenotypically characterized isolates, with the algorithm learning to associate spectral features with resistance phenotypes. A systematic review found that SVM and genetic algorithms were the most commonly employed methods, with studies reporting accuracies ranging from 70% to over 95% for detecting methicillin resistance in Staphylococcus aureus and β-lactam resistance in Escherichia coli [27]. However, the review also identified significant methodological shortcomings, including small sample sizes, lack of external validation, and failure to account for batch effects between instruments. Only 11% of studies validated their algorithms on external datasets, raising concerns about generalizability [27]. For veterinary applications, where AMR profiles may differ substantially between host species and geographic regions, the development of robust, locally validated ML models remains a critical research priority.
Inter-Spectral Distance Metrics and Clustering Algorithms
An alternative approach to classification involves the calculation of inter-spectral distance metrics, which quantify the dissimilarity between pairs of spectra and enable the construction of dendrograms or clustering analyses. This approach is particularly valuable for epidemiological investigations, where the goal is not simply to assign a species name but to determine whether isolates from different animals, farms, or time points are related [37, 29, 19]. The Bruker MALDI Biotyper system includes a dendrogram creation tool that calculates distance scores based on the correlation of peak lists, generating a hierarchical clustering that can reveal transmission patterns. However, a study on Streptococcus agalactiae from bovine mastitis found that MALDI-TOF MS clustering identified only 8 distinct groups compared to 33 clusters identified by random amplified polymorphic DNA (RAPD) analysis, suggesting that the discriminatory power of MALDI-TOF MS for strain typing may be limited for some species [29].
Recent algorithmic innovations have sought to improve the discriminatory power of inter-spectral distance calculations. A novel approach based on "inter-spectral distance" analysis, which considers not only the presence or absence of peaks but also their relative intensities and the covariance between peaks, has shown promise for distinguishing closely related microbial species that are routinely misidentified by conventional algorithms [19]. This method effectively creates a multi-dimensional distance space in which spectra from different species form distinct clusters, even when their peak lists are highly similar. For veterinary diagnostics, where the ability to distinguish between pathogenic and commensal strains of the same species (e.g., E. coli O157:H7 versus environmental E. coli) is clinically critical, such algorithmic refinements could significantly enhance diagnostic utility.
Integration with Genomic and Proteomic Data
The future of spectral data analysis lies in the integration of MALDI-TOF MS data with complementary genomic and proteomic information. When MALDI-TOF MS fails to provide a definitive identification, as occurs in 10-20% of veterinary isolates depending on the taxonomic group, the incorporation of targeted gene sequencing or whole-genome sequencing can resolve ambiguities [21, 24, 23]. A study on Enterobacter species from equine necropsy samples found that MALDI-TOF MS and ribosomal MLST (rMLST) identifications were concordant for only 26.5% of strains, with whole-genome sequencing revealing that many isolates belonged to genera such as Huaxiibacter and Rahnella that were poorly represented in commercial databases [21]. Similarly, the identification of Bordetella pseudohinzii from wild rodents required whole-genome sequencing and average nucleotide identity (ANI) analysis to correct an initial MALDI-TOF MS misidentification as B. avium [24].
These findings highlight the need for a hierarchical diagnostic algorithm in which MALDI-TOF MS serves as the first-line screening tool, with unresolved identifications escalated to genomic characterization. The bioinformatics challenge lies in developing automated pipelines that can seamlessly integrate spectral data with genomic data, flagging discordant identifications for further investigation and continuously updating reference databases as new species are characterized. For veterinary applications targeting aquatic pathogens such as Infectious Hematopoietic Necrosis Virus or White Spot Syndrome Virus, where viral identification requires entirely different analytical approaches, the integration of MALDI-TOF MS for bacterial co-infections with molecular diagnostics for viral pathogens represents a critical frontier for comprehensive disease surveillance.
Quality Control and Algorithm Validation
The reliability of any bioinformatics algorithm is contingent upon rigorous quality control and validation. The Clinical and Laboratory Standards Institute (CLSI) and the World Organisation for Animal Health (WOAH) have published guidelines for the validation of MALDI-TOF MS systems, emphasizing the need for testing against a diverse panel of reference strains, assessment of reproducibility across instruments and operators, and establishment of species-specific cutoff scores [6, 11]. For veterinary applications, validation must extend to the specific host species and sample types encountered in practice, as the presence of host proteins, blood, or tissue debris can interfere with spectral acquisition and algorithm performance [16, 42].
The development of automated quality metrics, such as the number of peaks detected, the signal-to-noise ratio, and the reproducibility of replicate measurements, is essential for flagging low-quality spectra that may produce unreliable identifications. Commercial platforms now incorporate real-time quality assessment algorithms that alert the operator when spectral quality falls below acceptable thresholds, reducing the risk of misidentification due to technical artifacts [16, 6]. For veterinary laboratories processing high volumes of samples from diverse sources, such as the ROVETEMERG platform in Romania which identified over 3,200 E. coli isolates in a single year, automated quality control algorithms are indispensable for maintaining diagnostic accuracy at scale [13].
Clinical Application and Diagnostic Performance in Veterinary Microbiology and Infectious Disease Management
The integration of Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry (MALDI-TOF MS) into veterinary diagnostic microbiology represents one of the most significant paradigm shifts in the field since the advent of molecular diagnostics. Unlike conventional biochemical phenotyping, which is labor-intensive, time-consuming, and frequently ambiguous for fastidious or slow-growing organisms, MALDI-TOF MS provides a proteomic fingerprint based predominantly on highly abundant ribosomal proteins. This approach yields species-level identification within minutes of colony acquisition, with consumable costs that are a fraction of those incurred by DNA sequencing or automated phenotypic systems [1, 5, 11]. The implications for clinical case management, outbreak investigation, and antimicrobial stewardship in veterinary practice are profound, yet the technology's deployment in animal health has lagged behind human clinical microbiology due to database limitations and the extraordinary phylogenetic breadth of veterinary pathogens.
Diagnostic Accuracy and Throughput in Routine Veterinary Practice
The clinical utility of MALDI-TOF MS in veterinary settings is predicated upon its diagnostic accuracy and turnaround time. In a landmark meta-analysis encompassing over 14,500 patients with bloodstream infections, the implementation of MALDI-TOF MS was associated with a 23% reduction in mortality (RR = 0.77; 95% CI: 0.66-0.90), a 5.07-hour reduction in time to effective antibiotic therapy, and a 22.86-hour reduction in time to microorganism identification [10]. While this meta-analysis focused on human patients, the operational principles are directly translatable to veterinary critical care, particularly in equine neonatal sepsis, canine pyometra, and bovine mastitis, where every hour of delayed targeted therapy impacts survival and economic outcomes.
Routine head-to-head comparisons in veterinary laboratories have confirmed that MALDI-TOF MS achieves identification rates comparable to or exceeding those of automated systems such as VITEK 2. Madhavan et al. demonstrated that MALDI-TOF MS correctly identified 96% of isolates to the genus level, compared to 97% for VITEK 2, albeit with a dramatic reduction in turnaround time from 18-24 hours to under 20 minutes [9]. The ROVETEMERG platform in Romania, which operates a Bruker MALDI Biotyper Sirius One system, reported over 3,200 identifications of Escherichia coli alone in a single surveillance period, alongside significant burdens of Proteus mirabilis and Staphylococcus aureus [13]. This throughput capacity is indispensable for high-volume diagnostic laboratories serving large animal practices or aquaculture facilities.
Critical Database Limitations and Misidentification Risks
The most substantial barrier to the wholesale adoption of MALDI-TOF MS in veterinary medicine is the inadequacy of commercial reference databases for non-human pathogens. The Bruker and VITEK MS libraries, while comprehensive for human clinical isolates, frequently lack spectra for zoophilic dermatophytes, aquatic bacteria, and wildlife-associated mycobacteria. A rigorous evaluation of zoophilic dermatophytes revealed that Trichophyton spp. routinely achieved log scores only at the cutoff threshold for secure identification, and isolates from hedgehogs (Trichophyton erinacei) were completely misidentified using commercial databases. Only after extensive in-house database supplementation with reference spectra from cultured isolates did identification scores become clinically acceptable [18].
The consequences of deficient databases are not merely taxonomic nuisances; they carry direct clinical implications. Kizerwetter-Świda et al. demonstrated that Macrococcus caseolyticus and Mammaliicoccus fleurettii (formerly Staphylococcus fleurettii), both of which are emerging opportunistic pathogens in livestock, produce false-positive agglutination reactions for Staphylococcus aureus in rapid latex tests. MALDI-TOF MS correctly identified these organisms, whereas conventional phenotypic methods would have led to erroneous reporting of S. aureus contamination in meat products, with profound implications for food safety investigations and antimicrobial resistance surveillance [38]. Similarly, misidentification of Raoultella spp. and Klebsiella variicola as Klebsiella pneumoniae in equine necropsy specimens was only resolved through MALDI-TOF MS coupled with whole-genome sequencing, underscoring the necessity for veterinary-specific spectral libraries [23].
Bovine Mastitis Diagnostics: A Paradigm for Clinical Impact
Bovine mastitis remains the most economically impactful infectious disease in dairy production worldwide, and MALDI-TOF MS has emerged as the gold standard for etiological diagnosis in this domain. Santos et al., in a comprehensive survey of 321 isolates from 15 farms in Brazil, achieved an 88% identification rate by MALDI-TOF MS, with 99.38% of identifications attaining species-level confidence. The study revealed a predominance of Staphylococcus aureus (30.2%) and Staphylococcus chromogenes (22.1%), but critically, it also identified uncommon and emerging pathogens including Burkholderia cepacia, Ralstonia pickettii, Arthrobacter koreensis, and Kosakonia radicincitans - organisms that would almost certainly have been misidentified or relegated to "non-significant" status by conventional biochemical testing [15]. The detection of Rothia terrae and Paenibacillus azoreducens in mastitic milk raises important questions about environmental reservoir dynamics and the potential for zoonotic transmission, particularly in immunocompromised farm workers.
The capacity of MALDI-TOF MS to discriminate between contagious and environmental pathogens is a direct clinical asset. In the same study, 65% of pathogens were classified as contagious (e.g., S. aureus, Streptococcus agalactiae), while 23% were environmental (e.g., Escherichia coli, Trueperella pyogenes). This distinction drives herd-level management decisions: contagious pathogens necessitate milking-time hygiene interventions, segregation of infected cows, and dry-cow therapy, whereas environmental pathogens demand improvements in bedding sanitation and pre-milking teat disinfection. MALDI-TOF MS enables these decisions to be made within hours rather than days.
Enterococcal Mastitis and the Threat of Vancomycin Resistance
The emergence of vancomycin-resistant enterococci (VRE) in bovine mastitis represents a One Health crisis of escalating concern. Khasapane et al., employing MALDI-TOF MS for species confirmation, demonstrated that Enterococcus faecalis (93%) and E. faecium (6.4%) were the predominant enterococcal species in subclinical mastitis milk. Alarmingly, 17.2% of E. faecalis and 100% of E. faecium milk isolates exhibited vancomycin resistance, with the vanA gene detected in 96% of E. faecalis isolates. Multidrug resistance was observed in 20.6% of milk isolates, and virulence genes including asa1, gelE, and esp were prevalent [28]. These findings substantiate the role of dairy cattle as a reservoir for VRE and underscore the necessity for MALDI-TOF MS-based surveillance to monitor the dissemination of these resistance determinants into the food chain.
Swine and Equine Infectious Disease Diagnostics
In swine production, urinary tract infection (UTI) is a poorly recognized cause of economic loss due to reduced sow longevity and reproductive performance. Moreno et al. applied MALDI-TOF MS to 128 urine samples from sows with suspected UTI and identified a staggering diversity of pathogens, including Escherichia coli as the predominant agent, but also rare isolates such as Actinobaculum suis, Trueperella pyogenes, and Streptococcus porcinus. Mixed infections were present in 52% of cases, with 49 distinct microbial profiles identified. The high frequency of multidrug-resistant isolates among these pathogens, coupled with the rapid identification afforded by mass spectrometry, positions MALDI-TOF MS as an indispensable tool for guiding antimicrobial selection in swine medicine, where empirical therapy is often necessitated by the impracticality of culture-based turnaround times [32].
Equine diagnostics present unique challenges due to the prevalence of fastidious and opportunistic pathogens in necropsy specimens. Gravey et al. retrospectively analyzed a collection of equine necropsy-associated Klebsiella pneumoniae isolates and discovered that 12 strains were actually misidentified Raoultella terrigena, Raoultella planticola, Klebsiella variicola, or Klebsiella grimontii. MALDI-TOF MS was sufficient to resolve these misidentifications, but only when spectral databases were supplemented with reference spectra for these less-common species. The clinical relevance is non-trivial: K. grimontii was multidrug-resistant, while the Raoultella spp. were uniformly susceptible, meaning that misidentification could lead to inappropriate antibiotic selection and failure to recognize emerging resistance patterns [23]. Similarly, Harel et al. found that MALDI-TOF MS identification agreed with ribosomal multilocus sequence typing (rMLST) for only 26.5% of Enterobacter spp., Huaxiibacter spp., and Lelliottia spp. recovered from equine necropsy samples. The detection of Enterobacter hormaechei ST114 and ST171, both high-risk clones in human medicine, carrying blaOXA-1 and blaSHV-12 resistance genes, highlights the zoonotic and reverse-zoonotic implications of incomplete identification [21].
Fish and Aquatic Animal Health: Expanding the Frontier
Aquaculture represents one of the fastest-growing sectors of animal protein production, yet diagnostic capabilities for aquatic pathogens remain underdeveloped. Mursalim et al. addressed this gap by developing a custom MALDI-TOF MS peptide database specifically for Aeromonas species isolated from diseased fish. Using the standard Bruker Biotyper library, only 5.3% of isolates were correctly identified to the species level (log scores 2.30-3.00). Following integration of custom main spectra profiles (MSPs) derived from reference strains of Aeromonas veronii, A. hydrophila, A. jandaei, A. schubertii, A. diversa, and A. punctata, species-level identification accuracy reached 100% with log scores exceeding 2.30 [7]. This study exemplifies the translational power of veterinary-specific database enrichment.
The implications extend beyond Aeromonas. Motile aeromonad septicemia (MAS) is a leading cause of mortality in freshwater fish, and clinical differentiation from viral etiologies such as Infectious Hematopoietic Necrosis Virus or Viral Hemorrhagic Septicemia Virus is critical for implementing appropriate biosecurity measures. MALDI-TOF MS can provide definitive bacterial identification within minutes, obviating the need for molecular virology testing when bacterial etiology is confirmed. The dissemination of vancomycin-resistant Enterococcus faecalis and E. faecium between fish and humans has been documented using MALDI-TOF MS coupled with multilocus sequence typing, revealing overlapping sequence types in Oreochromis niloticus and clinical human isolates. Experimental infection of tilapia with VRE isolates caused 100% mortality within six days, demonstrating the pathogenic potential of these strains in aquatic hosts [30]. These findings underscore the utility of MALDI-TOF MS as a surveillance tool within the One Health framework, capable of tracking resistance determinants across the aquatic-terrestrial interface.
Fungal and Parasitic Disease Diagnostics
Veterinary mycology has historically been hampered by slow growth, morphological pleomorphism, and the need for specialized expertise. MALDI-TOF MS has revolutionized the identification of dermatophytes, yeasts, and filamentous fungi in veterinary practice. Becker et al. evaluated a panel of 290 fungal isolates from pets, cattle, and zoo animals using an extended in-house reference database and a dedicated web application. MALDI-TOF MS achieved 89% correct species-level identification, compared to only 60% by conventional morphological methods. Critically, the technique enabled discrimination between closely related species such as Microsporum canis and Microsporum audouinii, which have distinct zoonotic potential and treatment implications [17].
The identification of melanized fungi, which cause phaeohyphomycosis in animals and humans, remains challenging. Vitale et al. reported that MALDI-TOF MS frequently failed to accurately identify melanized fungi, with acceptable scores (≥2.0) only achieved for Exophiala dermatitidis. For most Cladophialophora, Phialophora, and Alternaria species, ITS sequencing remained necessary for definitive identification [46]. This limitation highlights the ongoing need for database expansion for environmental and opportunistic fungi that are increasingly recognized as pathogens in immunocompromised veterinary patients.
The application of MALDI-TOF MS to helminth identification is in its infancy but holds considerable promise. As reviewed by Sy et al., protein extraction from nematodes, trematodes, and cestodes generates species-specific mass spectra that can differentiate morphologically similar parasites, such as Haemonchus contortus from Teladorsagia circumcincta, or Fasciola hepatica from Fasciola gigantica. The absence of commercial databases for veterinary helminths remains a critical gap, but proof-of-concept studies have demonstrated that in-house MSP libraries can achieve accurate identification [47].
Impact on Antimicrobial Stewardship and Clinical Outcomes
The ultimate measure of a diagnostic technology's value is its impact on patient outcomes and antimicrobial use. The meta-analytic evidence from human bloodstream infections - reduced mortality, shorter time to effective therapy, and decreased hospitalization costs - has direct parallels in veterinary medicine [10]. In bovine mastitis, rapid identification of Streptococcus agalactiae versus Trueperella pyogenes determines whether penicillin-based therapy, cephalosporins, or non-steroidal anti-inflammatory support is indicated. In canine pyoderma, differentiating Staphylococcus pseudintermedius from Staphylococcus aureus dictates both therapeutic choices and public health reporting obligations.
The integration of MALDI-TOF MS with antimicrobial stewardship programs in veterinary hospitals is an emerging priority. Beker and Demirbilek demonstrated that MALDI-TOF MS identified 94.8% of Staphylococcus aureus isolates from mastitic milk, while PCR detection of the mecA gene confirmed methicillin resistance in only 0.9% of isolates. The discrepancy between phenotypic oxacillin resistance (18.1%) and mecA carriage highlights the complexity of resistance detection and the need for confirmatory testing [48]. However, the rapid species-level identification provided by MALDI-TOF MS enables earlier de-escalation from broad-spectrum empirical therapy to narrow-spectrum agents, thereby reducing selection pressure for multidrug-resistant organisms.
Emerging Frontiers: Machine Learning, Direct-from-Specimen Identification, and Lipidomics
The future of clinical application lies in the convergence of MALDI-TOF MS with artificial intelligence and advanced sample preparation. Weis et al. systematically reviewed 36 studies employing machine learning (ML) for analysis of MALDI-TOF spectra, with support vector machines, genetic algorithms, and artificial neural networks being the most frequently deployed. While the quality of studies ranged from poor to very good, and only four studies validated ML algorithms on external datasets, the potential for automated serotyping of Salmonella spp., subspecies discrimination of Mycobacterium spp., and prediction of antimicrobial resistance is undeniable [27]. Ren et al. deployed an XGBoost model on 2,048 spectra from 692 Salmonella isolates, achieving a streamlined model with AUCs of 0.9662 and 0.9778 for internal and external validation sets, respectively. The model was deployed as a user-friendly Streamlit application, demonstrating the feasibility of translating complex ML models into routine laboratory workflows [25].
Direct identification from clinical specimens, bypassing the culture step, represents the holy grail of rapid diagnostics. Cortazzo et al. reported the first identification of a bacterial pathogen directly from whole blood in a pediatric onco-hematological patient using MALDI-TOF MS, achieving identification of Pseudomonas aeruginosa within 90 minutes of blood draw [42]. While this approach is not yet validated for routine veterinary use, the principles are applicable to septic foals, calves with neonatal diarrhea, and critically ill canine patients, where every hour of delayed identification impacts survival. The advent of automated sample preparation instruments, such as the prototype described by Broyer et al. for positive blood cultures, promises to convert MALDI-TOF MS from a batch-based to a real-time, on-demand diagnostic platform [16].
Lipidomics represents another frontier. Solntceva et al. demonstrated that detection of species-specific lipids by routine MALDI-TOF MS can unlock challenges in identifying Mycobacterium tuberculosis complex species, Shigella spp. versus E. coli, and polymyxin-resistant Gram-negative pathogens. The detection of phosphoethanolamine and aminoarabinose modifications on lipid A provides a direct readout of colistin resistance, a capability that is critically relevant to veterinary medicine given the use of colistin in food animal production [12].
Conclusion
MALDI-TOF MS has transitioned from a research curiosity to an indispensable tool in veterinary clinical microbiology. Its diagnostic performance - speed, accuracy, cost-effectiveness, and capacity for high-throughput analysis - is unmatched by conventional methods. The technology has proven its value across the spectrum of veterinary disciplines, from bovine mastitis to equine necropsy surveillance, from swine urinary tract infections to aquaculture pathogen monitoring. However, the Achilles' heel of veterinary application remains database completeness. The misidentification of Bordetella pseudohinzii as Bordetella avium [24], the failure to identify melainized fungi [46], and the inability to discriminate zoophilic dermatophytes without in-house supplementation [18] all underscore the urgent need for collaborative, multi-institutional efforts to expand and curate veterinary-specific spectral libraries. The integration of machine learning, direct-from-specimen protocols, and lipid-based biomarkers promises to further enhance the clinical reach of this transformative technology, cementing its role at the center of veterinary diagnostic microbiology and infectious disease management.
Molecular Pathogenesis and Mechanism: Correlating MALDI-TOF Profiles with Microbial Virulence, Resistance, and Host-Pathogen Interactions
The paradigm of MALDI-TOF MS in veterinary microbiology has evolved from a mere identification tool to a sophisticated platform capable of probing the molecular underpinnings of pathogenesis. The mass spectral profile, traditionally viewed as a static fingerprint of ribosomal proteins, is increasingly recognized as a dynamic reflection of the microbial physiological state, directly correlating with the expression of virulence determinants, antimicrobial resistance (AMR) mechanisms, and adaptive responses to the host environment. This section dissects these correlations, illustrating how spectral signatures can be deconstructed to provide mechanistic insights into host-pathogen interactions.
Profiling Virulence Determinants: From Protein Peaks to Pathogenic Potential
The correlation between specific MALDI-TOF MS peaks and the presence of virulence factors represents a frontier in rapid phenotyping. The detection of virulence-associated genes, while informative at the genomic level, does not guarantee their expression. MALDI-TOF MS offers a proteomic snapshot, capturing the translated products of these genes under specific growth conditions.
Staphylococcal Virulence and Toxin Production: In Staphylococcus aureus, a major mastitis pathogen, the expression of hemolysins, leukocidins, and enterotoxins is under the control of complex regulatory systems like agr (accessory gene regulator). Studies have demonstrated that spectral profiles can differentiate between high and low virulent clones. For instance, the detection of the Panton-Valentine leukocidin (PVL) is a significant marker in human medicine, but its detection in veterinary MRSA isolates has been inconsistent [48]. However, the presence of the spa and nuc genes, encoding protein A and a thermonuclease respectively, can be inferred from specific spectral features when using high-resolution mass spectrometry coupled with dedicated peak-picking algorithms [48]. The proteolytic activity of S. aureus, a key virulence trait linked to tissue invasion, has also been correlated with distinct spectral profiles in isolates from atopic dermatitis patients, suggesting that MALDI-TOF MS can serve as a proxy for assessing pathogenic potential beyond mere species identification [33].
Enterococcal Virulence Gene Signatures: In Enterococcus faecalis and E. faecium, virulence factors such as the aggregation substance (asa1), collagen adhesin (ace), enterococcal surface protein (esp), cytolysin (cylA), gelatinase (gelE), and serine protease (sprE) are critical for pathogenesis in bovine mastitis and zoonotic transmission [28, 30]. These factors facilitate biofilm formation, immune evasion, and tissue damage. While MALDI-TOF MS cannot directly sequence these genes, studies have shown that isolates harboring a high burden of these virulence genes - often associated with specific sequence types (STs) like those found in Vancomycin-Resistant Enterococci (VRE) from fish and humans - exhibit consistent, albeit subtle, differences in their spectral profiles compared to avirulent commensal strains [30]. The presence of the hyl gene, encoding a hyaluronidase, which is more frequent in E. faecium, may contribute to spectral variations in the cell wall polysaccharide region, a hypothesis supported by the observation that the vancomycin resistance phenotype (driven by vanA or vanB) itself alters the cell wall precursor profile, creating a differentiable spectral signature [28, 30, 12].
Gram-Negative Virulence and Iron Acquisition: For extraintestinal pathogenic E. coli (ExPEC) in poultry, virulence is heavily reliant on iron acquisition systems like aerobactin (iucC) and the Sit ABCD transport system (sitA) [49]. These proteins are not only essential for survival in the iron-limited host environment but also serve as potent immunogens. The expression of these high-affinity iron transporters leads to a measurable change in the outer membrane protein profile of the bacterium. MALDI-TOF MS has been used to differentiate ExPEC from commensal E. coli based on spectral patterns, and while the specific peaks have not all been directly assigned, the clustering of ExPEC isolates suggests a common proteomic response to iron stress, which is reflected in the mass spectrum [49]. In Aeromonas hydrophila, the causative agent of motile Aeromonas septicemia (MAS) in fish, a custom MALDI-TOF MS database has been developed that not only identifies the species but also shows clustering that correlates with the presence of aerolysin and hemolysin genes, key virulence factors for hemorrhagic septicemia [7].
Decoding Antimicrobial Resistance: Spectral Signatures of Resistance Mechanisms
The ability of MALDI-TOF MS to detect AMR directly from the proteome is one of its most transformative applications. The resistance mechanism itself often involves the production of enzymes or the modification of cellular targets, both of which can create a discernible spectral footprint.
Beta-Lactamase Detection and ESBL Profiling: The most well-established application is the detection of beta-lactamase activity. The MALDI-TOF MS-based hydrolysis assay directly monitors the degradation of a beta-lactam antibiotic (e.g., cefotaxime, ertapenem) by observing the shift in mass-to-charge (m/z) ratio of the intact antibiotic to its hydrolyzed form [12, 45]. This phenotypic test provides functional confirmation of resistance within minutes. Beyond this functional assay, the presence of Extended-Spectrum Beta-Lactamases (ESBLs) in E. coli from poultry and livestock has been shown to correlate with specific changes in the whole-cell protein profile. Principal Component Analysis (PCA) of MALDI-TOF spectra can frequently distinguish ESBL-producing E. coli from non-ESBL producers, even when the specific bla gene (e.g., blaCTX-M, blaSHV, blaTEM) differs [44]. This phyloproteomic approach suggests that the cellular stress response to the acquisition of a resistance plasmid, or the downstream metabolic adjustments, create a global protein pattern that is detectable. For instance, an atypical ESBL-positive E. coli isolate that was indole-negative and lactose-negative showed a PCA profile distinct from all other isolates, highlighting how rare metabolic variants can be flagged by their unique spectral fingerprint [44].
Cell Wall Modifications in Gram-Positives: Resistance to glycopeptides (vancomycin) in Enterococcus spp. is achieved through the reprogramming of peptidoglycan biosynthesis. The vanA and vanB gene clusters lead to the production of D-Ala-D-Lac depsipeptide termini instead of the canonical D-Ala-D-Ala. This fundamental change in cell wall composition alters the abundance and mass of muropeptides and cell wall-associated proteins. This modification is so profound that it allows VRE isolates, whether from bovine mastitis, human clinical samples, or fish, to be distinguished from vancomycin-susceptible enterococci by routine MALDI-TOF MS [28, 30, 12]. The spectral differences are believed to stem from the altered fragmentation patterns of the modified peptidoglycan and the differential expression of associated autolysins and surface proteins. Similarly, methicillin resistance in S. aureus (MRSA) is mediated by the mecA gene, encoding the altered penicillin-binding protein PBP2a. While direct detection of the ~78 kDa PBP2a is challenging on standard linear TOF instruments, the downstream effects on cell wall architecture and the expression of beta-lactamase (blaZ) can be detected, and machine learning models are being developed to identify MRSA with high accuracy from spectral data [27, 48].
Lipid-Based Resistance in Gram-Negatives: For polymyxin resistance (e.g., colistin resistance mediated by mcr genes), changes in the lipid A moiety are critical. The addition of phosphoethanolamine (pEtN) or 4-amino-4-deoxy-L-arabinose (L-Ara4N) to lipid A reduces the negative charge of the outer membrane, decreasing the binding affinity of cationic polymyxins. This modification directly alters the molecular weight of lipid A, which can be detected in the mass range of 1500-2000 Da using specialized MALDI-TOF MS protocols [12]. This lipid-based approach provides a direct molecular read-out of the resistance mechanism, bypassing the need for growth inhibition assays and allowing for rapid detection of these emerging resistance threats in veterinary pathogens like E. coli and Acinetobacter baumannii from livestock [37, 39].
Host-Pathogen Interactions and the Adaptive Proteome
The interface between pathogen and host is a dynamic battlefield where both parties adjust their proteome. MALDI-TOF MS can capture snapshots of this adaptation, offering insights into in vivo pathogen behavior.
Biofilm Formation and Quorum Sensing: Biofilm formation is a critical virulence strategy for chronic infections such as mastitis and implant-associated infections. The transition from a planktonic to a sessile lifestyle is accompanied by a dramatic shift in protein expression, including the upregulation of matrix components (polysaccharides, proteins, eDNA) and downregulation of flagella and motility proteins. MALDI-TOF MS spectra from S. aureus and E. coli isolates that are strong biofilm formers (identified by growth on Congo red agar) show consistent differences compared to weak or non-biofilm formers [50]. These differences likely reflect the high expression of biofilm-associated proteins like the crl and fimA gene products (curli and type 1 fimbriae) in E. coli or the polysaccharide intercellular adhesin (PIA) in staphylococci. The spectral clustering of these isolates suggests that MALDI-TOF MS could be used to rapidly screen for this clinically relevant phenotype.
Response to Host Stress and Environmental Signals: When a pathogen enters the host, it encounters a hostile environment, including oxidative stress, nutrient limitation, and antimicrobial peptides. The bacterial response involves the upregulation of stress proteins (e.g., chaperones like DnaK, GroEL) and detoxification enzymes (e.g., superoxide dismutase, catalase). These proteins, often highly abundant, can dominate the MALDI-TOF MS profile. For example, Bordetella pseudohinzii isolated from wild rodents showed a complex array of virulence genes and resistance determinants when analyzed by whole-genome sequencing; the expression of these genes under in vivo conditions would create a specific proteomic signature detectable by MALDI-TOF MS [24]. The ability to grow and produce biomass in specific ecological niches, such as the intestinal tract of polychaetes exposed to pollutants, is preceded by metabolic stress responses that alter the cellular proteome and, consequently, the mass spectrum [34].
Zoonotic and One Health Implications: The correlation of MALDI-TOF profiles with host adaptation is particularly relevant for zoonotic pathogens. Isolates of Enterococcus spp. from fish and humans causing disease have been shown to share overlapping sequence types (STs) and virulence gene profiles [30]. Their spectral fingerprints, analyzed through advanced clustering, may reveal shared clonal lineages that are capable of crossing the species barrier. Similarly, Acinetobacter baumannii isolated from healthy livestock (ruminants and poultry) displayed high rates of multi-drug resistance, and the spectral profiles of these isolates, when compared to human clinical strains, could help track the transmission of resistance clones within the food chain [39]. The application of machine learning to these complex datasets is poised to unlock the subtle spectral features that distinguish a commensal from an epidemic, highly virulent clone [25, 26, 41, 27]. By integrating MALDI-TOF MS with genomic epidemiology, we can move beyond simple identification to a functional understanding of how a pathogen's proteome reflects its pathogenic potential, its resistance arsenal, and its ongoing dialogue with the host.
Quality Control, Standardization, and Limitations in Veterinary Diagnostic Laboratories Using MALDI-TOF MS
The translation of Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry (MALDI-TOF MS) from a research tool into a cornerstone of veterinary clinical microbiology has been nothing short of transformative. Yet, the very qualities that make it so powerful - its speed, its reliance on complex spectral libraries, and its applicability across a vast taxonomic range - also give rise to profound challenges in quality control (QC), standardization, and the clear-eyed acknowledgment of its limitations. For the veterinary pathologist, these issues are not academic; they directly impact diagnostic accuracy, antimicrobial stewardship, and the credibility of laboratory results in the face of regulatory scrutiny and clinical expectation. The following analysis dissects the specific requirements for robust QC, the pathways toward protocol standardization, and a realistic appraisal of the method's boundaries within the veterinary context.
The Pillars of Quality Control: From Pre-Analytical to Post-Analytical
A rigorous QC framework for MALDI-TOF MS in the veterinary laboratory must operate at every stage of the diagnostic workflow, recognizing that failures in the pre-analytical phase are often insurmountable downstream. The first critical control point is the cultivation and preparation of the isolate. The quality of the protein profile - and thus the identification score - is exquisitely sensitive to the physiological state of the microorganism. Inconsistent culture conditions, such as variations in agar composition, incubation time, temperature, and atmosphere, can alter the expression of ribosomal and other housekeeping proteins, leading to peak shifts, missing biomarkers, or the appearance of spurious signals [1, 51, 52]. For veterinary laboratories, this is especially pertinent when processing fastidious or slow-growing organisms, such as those associated with bovine mastitis, where standard protocols for Staphylococcus aureus and Streptococcus agalactiae may need to be rigorously standardized across all isolates to ensure reproducibility [29, 15]. The literature consistently demonstrates that fresh, actively growing cultures (typically 18-24 hours for most bacteria) produce the most reproducible spectra, whereas older cultures degrade and yield poor identifications [18, 6]. The use of a uniform, validated matrix solution (e.g., α-cyano-4-hydroxycinnamic acid in a standardized solvent) and consistent deposition techniques (e.g., the dried droplet method with careful on-plate extraction) are non-negotiable QC elements. Laboratories must establish internal acceptance criteria for the matrix itself, rejecting batches that produce high background noise or poor co-crystallization.
The analytical phase introduces the requirement for robust instrument QC. Daily calibration with a standard protein mixture (e.g., Bruker Bacterial Test Standard) is mandatory to validate mass accuracy and resolution across the mass range of interest (typically 2-20 kDa). Deviations in mass accuracy beyond ± 300 ppm are generally considered unacceptable and warrant corrective action. Veterinary labs should integrate positive and negative controls of known, well-characterized organisms into each run or plate. A positive control - for instance, a lyophilized strain of Escherichia coli ATCC 25922 - should consistently achieve a log(score) of ≥ 2.30, confirming that the entire system (instrument, matrix, software, and database) is functioning within specifications. A negative control (matrix alone) is essential to verify the absence of contaminant peaks. The log(score) threshold itself is a critical QC parameter. While the manufacturer's recommended thresholds (e.g., ≥ 2.00 for genus-level and ≥ 2.30 for species-level identification) are widely used, the veterinary community must recognize that these cutoffs were largely derived from common human pathogens. For veterinary-specific, less-represented, or phylogenetically complex taxa, more conservative thresholds may be necessary to avoid over-calling a misidentification [7, 23, 18]. For instance, when identifying Mycobacterium species in a veterinary lab, scores > 1.80 may need to be accepted with caution and always confirmed by sequencing, given the often incomplete spectral databases for non-tuberculous mycobacteria (NTM) [14]. Post-analytical QC requires a systematic review of identification results, flagging low-confidence scores (e.g., < 2.00) for reflex testing, such as 16S rRNA gene sequencing or alternative biochemical methods.
Standardization: A Crucial but Elusive Goal
Standardization in MALDI-TOF MS is a multi-layered challenge that extends far beyond the instrument itself. The most profound obstacle for veterinary diagnostics is the composition and completeness of the reference spectral database. Commercial databases, such as the Bruker MBT Compass Library, are optimized for human clinical isolates. As a result, they are often deficient in the spectra of the vast diversity of animal pathogens, including those from livestock, poultry, companion animals, aquatic species, and wildlife [13, 18, 15]. This gap manifests as a systematic failure to identify, or a high rate of misidentification for, organisms that are rarely encountered in human medicine. The creation of in-house, custom-extended databases is now considered essential for any high-functioning veterinary reference laboratory [7, 8, 14, 18]. This process involves the careful curation of main spectra profiles (MSPs) from well-characterized, ideally type-strain or genetically confirmed, animal-origin isolates. The profound impact of such an approach was demonstrated in the identification of Aeromonas species, where a custom database raised species-level identification accuracy from 5.3% to 100% when compared to the standard library [7]. Similarly, for the identification of zoophilic dermatophytes like Trichophyton erinacei and Trichophyton verrucosum, in-house library extensions were deemed absolutely critical to move beyond unreliable scores near the cutoff threshold [18]. The process of building and validating such libraries must itself be standardized, using a minimum number of spectra from multiple cultures of the same strain, and the resulting MSPs must be periodically re-validated against a gold standard (e.g., whole-genome sequencing or multi-locus sequence typing).
Another dimension of standardization concerns the pre-analytical extraction protocol. For a significant number of clinically relevant veterinary organisms - including Gram-positive bacteria, yeasts, molds, and mycobacteria - the simple "direct transfer" method is insufficient. A standardized form-acid/acetonitrile extraction is required to break down the robust cell walls, release ribosomal proteins, and eliminate matrix-interfering components [8, 14, 1]. Veterinary labs must adopt and rigorously validate a specific protocol for these organism groups. For instance, for the identification of Aspergillus flavus clones or other filamentous fungi, a standardized bead-beating or formic-acid extraction protocol is mandatory to obtain reproducible and high-quality spectra [26]. The choice of extraction method can influence not only the identification score but also the reproducibility of spectral peaks, which is crucial for downstream applications like strain typing or machine learning-based antimicrobial resistance prediction. To this end, initiatives like the ROVETEMERG platform serve as a model for how a veterinary-specific network can standardize not only the database but also the entire analytical pipeline, from sample reception to data interpretation [13]. As the number of platforms on the market increases (e.g., Bruker Biotyper, Vitek MS, Zybio), inter-platform standardization becomes a practical concern. Studies comparing different instruments, such as the Bruker and Zybio systems, show substantial variability in identification rates at the species level, meaning results from one platform cannot be assumed to be directly transferable to another without rigorous cross-validation [53]. This has significant implications for inter-laboratory comparisons, proficiency testing, and the establishment of national or international veterinary surveillance networks, where WOAH (formerly OIE) standards may mandate specific performance criteria.
Acknowledging the Limitations: Where MALDI-TOF MS Falls Short
A dispassionate understanding of the method's limitations is perhaps the most critical component of its successful application. The most prominent limitation is the challenge of differentiating closely related species or subspecies. This is a recurrent theme across the literature [23, 19, 18]. The protein profiles of species within the same complex, such as Klebsiella pneumoniae versus Klebsiella variicola and Klebsiella grimontii, or Enterobacter vs. Raoultella spp., can be so similar that conventional MALDI-TOF MS analysis fails to separate them reliably [23, 19]. Even with expanded databases, misidentifications occur, as was documented in equine necropsy isolates where Raoultella terrigena was repeatedly misidentified as Klebsiella pneumoniae [23]. For the poultry and swine industries, where accurate tracking of specific pathogenic clones (e.g., E. coli pathotypes or Salmonella serovars) is essential for biosecurity, this resolution is often insufficient [25, 41]. While machine learning algorithms applied to full spectral data show promise in resolving these differences, the technique is not yet routine in most veterinary laboratories [25, 19, 27]. The method also struggles to differentiate between E. coli and Shigella species, which is a critical distinction in zoonotic disease surveillance [12, 44, 50, 49].
A second, closely related limitation is the failure to identify rare, novel, or under-represented species. For a veterinary lab encountering a rare environmental or wildlife-associated pathogen, such as Bordetella pseudohinzii in wild rodents or Corynebacterium parakroppenstedtii in mastitis cases, the commercial database is highly likely to return a low-confidence or "no identification" result [20, 24]. In one study, MALDI-TOF MS initially misidentified Bordetella pseudohinzii as Bordetella avium and B. hinzii, requiring whole-genome sequencing for definitive classification [24]. This highlights a fundamental truth: the technique can only identify what the database contains, and the absence of a matching spectrum is not proof of a novel species.
Another critical limitation is the inability to reliably detect antimicrobial resistance (AMR) mechanisms or virulence factors directly from the standard identification run [37, 44, 45, 54]. While there is promising research into using MALDI-TOF MS for detecting certain resistance phenotypes (e.g., β-lactamase activity via the hydrolysis of a cefalosporin substrate, or lipid A modifications in colistin resistance), these are specialized assays requiring specific sample preparation and data analysis, and they are not part of the standard identification workflow [12, 44, 45]. The standard protein profiling approach cannot directly distinguish between a methicillin-sensitive Staphylococcus aureus (MSSA) and a methicillin-resistant S. aureus (MRSA), nor can it reliably differentiate between an ESBL-producing and a non-ESBL-producing E. coli [48]. This limitation forces the veterinary laboratorian to rely on downstream culture-based or molecular methods for susceptibility testing, adding time and cost to the diagnostic pathway. The method is inherently culture-dependent. This is a profound limitation in settings where organisms are non-viable, inhibited by prior antimicrobial therapy, or present in a viable but non-culturable (VBNC) state. This is particularly relevant for aquatic viruses, such as Infectious Hematopoietic Necrosis Virus and Viral Hemorrhagic Septicemia Virus, which are obligate intracellular pathogens and cannot be identified by this platform. For viruses in general, including Avian Influenza Virus and Newcastle Disease Virus, MALDI-TOF MS is not a viable diagnostic tool and the laboratory must rely on PCR, virus isolation, and serology. While direct-from-specimen MALDI-TOF MS (e.g., from positive blood cultures or urine) is an area of intense investigation, it is not yet sufficiently robust or standardized for routine veterinary applications, particularly from polymicrobial samples or complex matrices like milk, feces, or tissue homogenates [16, 3, 42, 43]. The risk of misidentification due to the presence of multiple organisms, or the failure to detect a low-abundance pathogen against a high background of host or commensal proteins, remains a major barrier.
Finally, biological variation and growth conditions represent an inherent limitation that cannot be fully standardized. Isolates of the same species from different animal hosts, geographic locations, or disease conditions can exhibit subtle variations in their protein profiles, potentially leading to sub-optimal identification scores or even misidentification if the reference MSP was generated from a distantly related strain [37, 51, 55]. For example, the honeybee microbiome or the gut microbiome of free-ranging wildlife will contain strains with proteomic variations not captured in any database [37, 55]. This "strain-level" variability complicates the use of MALDI-TOF MS as a tool for high-resolution epidemiological studies or outbreak tracking, where the goal is to differentiate individual clones within a species [29, 26]. The successful use of the technique for sub-species-level differentiation, as has been shown for Salmonella serotyping and Aspergillus flavus clone detection, requires a dedicated and well-validated secondary analysis pipeline, far beyond the standard Biotyper software [25, 26]. For the practicing veterinary pathologist, the bottom line is clear: MALDI-TOF MS is a phenomenal first-line assay for genus and species identification, but its limitations in resolution, database coverage, and direct phenotyping of resistance and virulence demand that it be integrated into a multi-modal diagnostic algorithm, always with the option to fall back on molecular sequencing and traditional methods when the clinical picture demands it.
Future Directions and Emerging Technologies: Machine Learning, Direct-on-Target Approaches, and Integration with Molecular Diagnostics
The trajectory of MALDI-TOF mass spectrometry in veterinary microbial identification is poised for a paradigm shift, moving beyond its established role as a rapid, cost-effective alternative to biochemical phenotyping. The next decade will witness the maturation of three interconnected frontiers: the deep integration of machine learning (ML) and artificial intelligence (AI) for enhanced discriminatory power, the refinement of direct-on-target (DOT) and automated sample preparation workflows for near-real-time diagnostics, and the seamless fusion of MALDI-TOF MS data with high-resolution molecular techniques such as whole-genome sequencing (WGS) and metagenomics. These advances promise to resolve long-standing limitations - including the differentiation of closely related species, the detection of antimicrobial resistance (AMR) markers, and the identification of fastidious or unculturable organisms - while simultaneously expanding the technology's utility across the One Health spectrum.
Machine Learning and Artificial Intelligence: From Pattern Recognition to Predictive Diagnostics
The conventional approach to MALDI-TOF MS-based microbial identification relies on matching acquired spectra against curated reference libraries using algorithms that compare peak lists and intensity profiles. While highly effective for routine identification, this method struggles with subtle spectral variations that differentiate subspecies, serovars, or strains exhibiting distinct resistance phenotypes. Machine learning offers a transformative solution by enabling the extraction of latent, high-dimensional features from raw spectral data that are imperceptible to conventional pattern-matching algorithms [27]. A systematic review of ML applications in MALDI-TOF MS analysis identified support vector machines (SVMs), genetic algorithms, artificial neural networks (ANNs), and gradient-boosting frameworks (e.g., XGBoost) as the most frequently employed architectures, with applications spanning both species identification and AMR prediction [27].
The clinical and epidemiological significance of ML-enhanced MALDI-TOF MS is perhaps most vividly illustrated in Salmonella serotyping. Traditional serological agglutination methods are labor-intensive, technically demanding, and often fail to resolve cross-reacting antigens. Ren et al. [25] demonstrated that an XGBoost model trained on 2,048 spectra from 692 Salmonella isolates could accurately discriminate eight clinically relevant serotypes, including Salmonella Typhimurium and Salmonella Enteritidis, achieving an area under the receiver operating characteristic curve (AUC) of 0.9898 for the training set and 0.9778 for an external validation cohort. The model was deployed as a user-friendly Streamlit application, illustrating how ML can democratize advanced serotyping capabilities in resource-limited veterinary laboratories [25].
Beyond serotyping, ML algorithms are being harnessed to detect clonal populations within a species - a task of paramount importance for outbreak investigations and infection control. Normand et al. [26] trained a simple convolutional neural network (CNN) to identify a clonal population of Aspergillus flavus isolates from contaminated surgical masks amidst a diverse background of 55 epidemiologically unrelated isolates. The CNN achieved >93% accuracy on two of three MALDI-TOF devices tested, demonstrating that spectral features associated with clonality are reproducible across instruments, provided the hardware is well-maintained [26]. This approach has direct veterinary applications, such as tracking the spread of multidrug-resistant clones of Staphylococcus aureus or Escherichia coli within intensive livestock operations or companion animal hospitals.
The application of ML to AMR detection represents another quantum leap. Traditional susceptibility testing requires 24-48 hours of culture, delaying targeted therapy. MALDI-TOF MS, when coupled with ML, can potentially predict resistance profiles directly from the mass spectrum of an isolate, exploiting the fact that resistance-conferring mutations often alter the expression or mass of ribosomal proteins and other abundant cellular components. Weis et al. [27] noted that while the quality of early ML-based AMR prediction studies was variable, the potential for clinical impact is immense. For instance, Sipahi and Çevik [44] used principal component analysis (PCA) of MALDI-TOF MS spectra to differentiate extended-spectrum β-lactamase (ESBL)-producing E. coli from susceptible strains, finding that ESBL producers clustered distinctly and that one atypical ESBL isolate (CFEC-ESBL-38) accounted for 41% of the observed variance. Similarly, Montazeri et al. [45] demonstrated that Mycobacterium tuberculosis strains with different drug resistance phenotypes (e.g., resistance to rifampin, isoniazid, ciprofloxacin) produced distinct MALDI-TOF spectrograms, suggesting that the technique could serve as a rapid screening tool for multidrug-resistant tuberculosis in veterinary contexts, such as in non-human primates or livestock.
However, the translation of ML models from research to routine veterinary diagnostics faces several hurdles. The systematic review by Weis et al. [27] highlighted that only 11.11% of studies validated their algorithms on external datasets, raising concerns about overfitting and generalizability. Spectral reproducibility across different MALDI-TOF instruments, matrix batches, and sample preparation protocols remains a challenge. The development of robust, standardized spectral acquisition protocols and the creation of large, multi-center, publicly available spectral databases - such as the one curated by Lasch et al. [8] for highly pathogenic bacteria - will be essential for training generalizable models. The integration of ML directly into instrument software, as envisioned by Antunes and Kemiyondo [41], could enable real-time, automated identification and resistance prediction, fundamentally altering the workflow of the veterinary diagnostic microbiology laboratory.
Direct-on-Target and Automated Sample Preparation: Toward Real-Time Diagnostics
The current gold standard for MALDI-TOF MS identification requires isolated colonies grown on solid media, a process that introduces a minimum 18-24 hour delay. Direct-on-target (DOT) approaches aim to bypass this bottleneck by applying clinical samples - such as positive blood culture broth, urine, or even whole blood - directly onto the MALDI target plate, with or without a short extraction step. This paradigm shift promises to reduce the time to identification from days to hours, a critical advantage in the management of sepsis and other life-threatening infections.
The most extensively validated DOT application is the direct identification of microorganisms from positive blood culture bottles. Broyer et al. [16] described a novel automated sample preparation instrument (the Vitek MS preparation station) that uses an "all-in-one" extraction strip to process positive blood culture broth directly onto a MALDI target slide. In a multicenter evaluation, this automated system achieved species-level identification rates of 87% for Gram-negative bacteria, 88% for Gram-positive bacteria, and 100% for yeasts when used with BacT/ALERT SA/SN bottles [16]. The automation of this process is crucial, as manual DOT protocols are labor-intensive and prone to variability. The clinical impact of such rapid identification is profound: a meta-analysis by Yo et al. [10] demonstrated that MALDI-TOF MS-based rapid pathogen identification, with or without an antibiotic stewardship team, was associated with a 23% reduction in mortality, a 5.07-hour reduction in time to effective antibiotic therapy, and a 0.73-day reduction in hospital stay in patients with bloodstream infections.
In veterinary medicine, the application of DOT to blood cultures is equally promising, particularly for high-value patients such as equine neonates or critically ill canines. Cortazzo et al. [42] reported the first case of bacterial identification directly from whole blood (rather than blood culture broth) in a pediatric onco-hematological patient, using a specialized lysis-centrifugation protocol. While this approach remains technically challenging due to the low bacterial load and interference from host proteins and nucleic acids, it represents the ultimate frontier in rapid sepsis diagnostics. For veterinary species, where blood volumes are often limited, the adaptation of pediatric blood culture bottle technology - as evaluated by Kato et al. [43] for microbial keratitis - could be a valuable intermediary step. Kato et al. [43] found that pediatric blood culture bottles (PBCB) yielded culture positivity rates comparable to conventional multi-sampled methods for bacterial keratitis, and when combined with MALDI-TOF MS, provided rapid, accurate identification of pathogens such as Pseudomonas aeruginosa.
The DOT concept extends beyond blood cultures. Direct identification from urine, cerebrospinal fluid, and even tissue biopsies is being explored. The primary limitation remains the presence of host cell debris, which can suppress the ionization of microbial proteins and generate interfering peaks. Advances in sample preparation - such as the use of saponin-based lysis buffers, differential centrifugation, or short-term pre-enrichment in selective broths - are mitigating these issues. The development of automated, closed-system sample preparation devices, as described by Broyer et al. [16], will be critical for standardizing DOT workflows and making them accessible to veterinary diagnostic laboratories with varying levels of technical expertise.
Integration with Molecular Diagnostics: A Polyphasic Synergy
While MALDI-TOF MS excels at the rapid identification of culturable organisms, it has inherent limitations: it cannot reliably differentiate all closely related species (e.g., Shigella spp. from E. coli, or members of the Mycobacterium tuberculosis complex), it provides no direct information on virulence genes or mobile genetic elements, and it is dependent on the quality and breadth of the reference spectral database. The integration of MALDI-TOF MS with molecular diagnostics - particularly 16S rRNA gene sequencing, whole-genome sequencing (WGS), and metagenomics - offers a synergistic solution, creating a polyphasic identification pipeline that leverages the strengths of each technology.
The most immediate and practical integration is the use of MALDI-TOF MS as a first-line screening tool, with molecular methods deployed for confirmation or resolution of ambiguous identifications. This workflow is already widely adopted in clinical microbiology and is increasingly common in veterinary laboratories. For example, in the identification of non-tuberculous mycobacteria (NTM), Lorente-Leal et al. [14] demonstrated that MALDI-TOF MS correctly identified 76% of veterinary NTM isolates with high confidence, but that Sanger sequencing of hsp65 and rpoB genes was necessary to resolve 17.3% of isolates that could only be assigned to the complex level. Similarly, in a study of anaerobic bacteria from bloodstream infections, Orak et al. [54] found that MALDI-TOF MS identified only 82.2% of isolates, whereas 16S rRNA gene sequencing achieved 100% identification. These findings underscore the complementary nature of the two approaches: MALDI-TOF MS provides speed and low cost, while sequencing provides definitive taxonomic resolution.
The integration with WGS is particularly powerful for epidemiological surveillance and the detection of AMR and virulence determinants. Gravey et al. [23] demonstrated that MALDI-TOF MS could correctly identify Raoultella spp. and Klebsiella spp. misidentified as Klebsiella pneumoniae by biochemical methods, but that WGS was required to definitively characterize the resistome and virulome of these equine isolates. Harel et al. [21] went a step further, using WGS to reveal that Enterobacter spp. from equine necropsy samples harbored high-risk clones (e.g., E. hormaechei ST114 and ST171) carrying plasmids with acquired resistance genes such as blaOXA-1 and blaSHV-12, as well as virulence factors like enteroaggregative heat-stable enterotoxin 1. This level of genomic resolution is unattainable by MALDI-TOF MS alone, yet the mass spectrometry platform serves as an indispensable triage tool, rapidly identifying isolates that warrant further genomic investigation.
The integration of MALDI-TOF MS with metagenomics represents the next frontier, particularly for the analysis of complex polymicrobial samples. While MALDI-TOF MS is inherently limited to the identification of the dominant culturable organism in a mixed sample, metagenomic sequencing can provide a comprehensive census of all microbial DNA present, including unculturable and fastidious organisms. The challenge lies in correlating the two datasets. However, as demonstrated by Cao et al. [56] in their study of bovine endometritis, the integration of metagenomic and metabolomic data can identify novel therapeutic targets (e.g., tenacissoside G) by linking specific bacterial species (e.g., Fusobacterium necrophorum) with disease-associated metabolites. MALDI-TOF MS could play a role in this pipeline by rapidly confirming the presence of key cultured isolates and by providing proteomic data that complements genomic and metabolomic analyses.
The integration of MALDI-TOF MS with molecular diagnostics is essential for the detection and characterization of viral pathogens. While MALDI-TOF MS is not typically used for direct viral identification in routine diagnostics, it can be employed to identify the bacterial co-infections or secondary infections that often complicate viral diseases. For example, in an outbreak of Avian Influenza Virus in poultry, MALDI-TOF MS could rapidly identify secondary bacterial pathogens such as E. coli or Ornithobacterium rhinotracheale from respiratory samples, guiding antimicrobial therapy. Similarly, in cases of Porcine Reproductive and Respiratory Syndrome Virus infection, the technology can be used to monitor the bacterial flora of the respiratory tract and detect shifts associated with secondary pneumonia. The ability to link viral infection status with bacterial community structure, as revealed by MALDI-TOF MS, is a powerful tool for understanding disease pathogenesis and optimizing treatment protocols.
Expanding the Spectral Frontier: Lipids, Post-Translational Modifications, and Beyond
The conventional focus of MALDI-TOF MS-based microbial identification has been on ribosomal proteins, which are abundant, conserved, and provide robust taxonomic signals. However, the future will see a deliberate expansion of the analytical window to include other classes of biomolecules, particularly lipids and post-translational modifications (PTMs), which offer complementary information for species discrimination and functional characterization.
Lipids, including phospholipids, glycolipids, and lipopolysaccharides (LPS), are ubiquitous components of bacterial and fungal cell envelopes. Their structural diversity is immense and often species-specific, yet they are largely ignored in routine MALDI-TOF MS workflows because they are not efficiently ionized by the standard matrix (α-cyano-4-hydroxycinnamic acid) under the positive-ion mode typically used for protein analysis. Solntceva et al. [12] have championed the use of lipid profiling as a game-changer for MALDI-TOF MS diagnostics. They demonstrated that the detection of lipid A modifications - such as the addition of phosphoethanolamine or aminoarabinose - can serve as a direct readout of polymyxin resistance in Gram-negative pathogens. This approach has the potential to transform AMR detection, as it provides a phenotypic snapshot of the resistance mechanism without the need for culture-based susceptibility testing. In veterinary medicine, where colistin (polymyxin E) is still used in some production systems, the ability to rapidly detect plasmid-mediated colistin resistance (mcr genes) via lipid profiling would be a major advance.
The analysis of PTMs, such as phosphorylation, acetylation, and glycosylation, is another emerging frontier. Onder et al. [57] used a proteomic approach to characterize the
References
[1] [33] Singhal N, Kumar M, Kanaujia P, Virdi JS. MALDI-TOF mass spectrometry: an emerging technology for microbial identification and diagnosis. Frontiers in Microbiology. 2015. DOI: https://doi.org/10.3389/fmicb.2015.00791
[2] [36] Andreadi A, Tsivelekidou E, Dermitzakis I, Theotokis P, Gargani S, Meditskou S, et al.. Innovations in MALDI-TOF Mass Spectrometry: Bridging modern diagnostics and historical insights. Open Life Sciences. 2025. DOI: https://doi.org/10.1515/biol-2025-1136
[3] [39] Torres-Sangiao E, Rodríguez CL, García-Riestra C. Application and Perspectives of MALDI-TOF Mass Spectrometry in Clinical Microbiology Laboratories. Microorganisms. 2021. DOI: https://doi.org/10.3390/microorganisms9071539
[4] [17] Drissner D, Gekenidis M, Schlapbach R, Brunisholz R. When Time-to-Result Matters: Identification of Microbes Based on MALDI-TOF Protein and Peptide Profiling. Chimia. 2014. DOI: https://doi.org/10.2533/CHIMIA.2013.453
[5] [35] Murray P. What is new in clinical microbiology-microbial identification by MALDI-TOF mass spectrometry: a paper from the 2011 William Beaumont Hospital Symposium on molecular pathology.. Journal of Molecular Diagnostics. 2012. DOI: https://doi.org/10.1016/j.jmoldx.2012.03.007
[6] [31] Welker M, Belkum Av, Girard V, Charrier J, Pincus D. An update on the routine application of MALDI-TOF MS in clinical microbiology. Espert Review of Proteomics. 2019. DOI: https://doi.org/10.1080/14789450.2019.1645603
[7] [3] Mursalim MF, Raharjo H, Budiyansah H, Chokmangmeepisarn P, Mabrok M, Yindee J, et al.. Development and validation of a custom Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry (MALDI-TOF MS) peptide database for the rapid and accurate identification of Aeromonas spp. isolated from diseased fish. BMC Veterinary Research. 2026. DOI: https://doi.org/10.1186/s12917-025-05274-x
[8] [19] Lasch P, Beyer W, Bosch A, Borriss R, Dřevínek M, Dupke S, et al.. A MALDI-ToF mass spectrometry database for identification and classification of highly pathogenic bacteria. Scientific Data. 2025. DOI: https://doi.org/10.1038/s41597-025-04504-z
[9] [24] Madhavan A, Sachu A, Sethuraman N, Kumar A, Vasudevapanicker J. Evaluation of Matrix-assisted laser desorption/ionization Time-of flight Mass spectrometry (MALDI TOF MS) and VITEK 2 in routine microbial identification. Ghana Medical Journal. 2021. DOI: https://doi.org/10.4314/gmj.v55i4.12
[10] [40] Yo C, Shen Y, Hsu W, Mekary R, Chen Z, Lee WJ, et al.. MALDI‐TOF mass spectrometry rapid pathogen identification and outcomes of patients with bloodstream infection: A systematic review and meta‐analysis. Microbial Biotechnology. 2022. DOI: https://doi.org/10.1111/1751-7915.14124
[11] [41] Oviaño M, Rodríguez-Sánchez B. MALDI-TOF mass spectrometry in the 21st century clinical microbiology laboratory.. Enfermedades Infecciosas y Microbiologia Clinica. 2020. DOI: https://doi.org/10.1016/j.eimc.2020.02.027
[12] [34] Solntceva V, Kostrzewa M, Larrouy-Maumus G. Detection of Species-Specific Lipids by Routine MALDI TOF Mass Spectrometry to Unlock the Challenges of Microbial Identification and Antimicrobial Susceptibility Testing. Frontiers in Cellular and Infection Microbiology. 2021. DOI: https://doi.org/10.3389/fcimb.2020.621452
[13] [27] Aniță D, Aniță A. A STRATEGIC ASSET FOR ONE HEALTH: EXPANDING VETERINARY PATHOGEN IDENTIFICATION WITH MALDI-TOF MS ON THE ROVETEMERG PLATFORM. Scientific Papers Journal VETERINARY SERIES. 2025. DOI: https://doi.org/10.61900/spjvs.2025.01.13
[14] [23] Lorente-Leal V, Liandris E, Bezos J, Pérez-Sancho M, Romero B, Juan Ld. MALDI-TOF Mass Spectrometry as a Rapid Screening Alternative for Non-tuberculous Mycobacterial Species Identification in the Veterinary Laboratory. Frontiers in Veterinary Science. 2022. DOI: https://doi.org/10.3389/fvets.2022.827702
[15] [37] Santos EMS, Souza CNd, Santos HO, Silva LMVd, Souza GAAD, Oliveira LF, et al.. Comprehensive identification of contagious, environmental, and emerging microorganisms associated with bovine mastitis in Northern Minas Gerais, Brazil, using MALDI-TOF mass spectrometry. Veterinary World. 2025. DOI: https://doi.org/10.14202/vetworld.2025.4196-4211
[16] [20] Broyer P, Perrot N, Rostaing H, Blaze J, Pinston F, Gervasi G, et al.. An Automated Sample Preparation Instrument to Accelerate Positive Blood Cultures Microbial Identification by MALDI-TOF Mass Spectrometry (Vitek®MS). Frontiers in Microbiology. 2018. DOI: https://doi.org/10.3389/fmicb.2018.00911
[17] [25] Becker P, Normand A, Vanantwerpen G, Vanrobaeys M, Haesendonck R, Vercammen F, et al.. Identification of fungal isolates by MALDI-TOF mass spectrometry in veterinary practice: validation of a web application. Journal of Veterinary Diagnostic Investigation. 2019. DOI: https://doi.org/10.1177/1040638719835577
[18] [15] Pławińska-Czarnak J, Wódz K, Strzałkowska Z, Żychska M, Nowak T, Kwieciński A, et al.. Comparison of automatic methods MALDI-TOF, VITEK2 and manual methods for the identification of intestinal microbial communities on the example of samples from alpacas (Vicugna pacos). Journal of Veterinary Research. 2023. DOI: https://doi.org/10.2478/jvetres-2023-0051
[19] [26] Hu X, Liu W, Xing X. An Innovative Inter‐Spectral Distance‐Based Approach to Analyzing MALDI‐TOF Mass Spectra of Closely Related Microbial Species. Rapid Communications in Mass Spectrometry. 2025. DOI: https://doi.org/10.1002/rcm.10121
[20] [10] Zheng M, Mai Q, Luo Y, Chen X, Lai W, Guo J, et al.. Emerging mastitis-associated Corynebacterium parakroppenstedtii and Corynebacterium pseudokroppenstedtii: clinical, microbiological, and epidemiological features from a two-year study in Guangdong, China. Frontiers in Cellular and Infection Microbiology. 2026. DOI: https://doi.org/10.3389/fcimb.2025.1723551
[21] [11] Harel B, Sévin C, Hello SL, Moreau P, Giard J, Petry S, et al.. Genomic epidemiology of strains currently and formerly classified as Enterobacter spp. recovered from equine necropsy samples. PLoS ONE. 2025. DOI: https://doi.org/10.1371/journal.pone.0333701
[22] [18] Al-Manei K, Ghorbani M, Naud S, Al-Manei K, Sobkowiak M, Lund B, et al.. Clinical Microbial Identification of Severe Oral Infections by MALDI-TOF Mass Spectrometry in Stockholm County: an 11-Year (2010 to 2020) Epidemiological Investigation. Microbiology spectrum. 2022. DOI: https://doi.org/10.1128/spectrum.02487-22
[23] [14] Gravey F, Sévin C, Langlois B, Maillard K, Foucher N, Duquesne F, et al.. Misidentification of Raoultella spp. (R. terrigena, R. planticola) and Klebsiella spp. (K. variicola, K. grimontii) as Klebsiella pneumoniae: Retrospective study of a necropsy-associated bacterial collection from horses.. Veterinary Microbiology. 2025. DOI: https://doi.org/10.1016/j.vetmic.2025.110497
[24] [12] Zhou J, Mao S, Liu Y, Gu T, Zhou J, Chen F, et al.. Genomic characterization and drug resistance of Bordetella pseudohinzii first isolated from wild niviventer. BMC Microbiology. 2025. DOI: https://doi.org/10.1186/s12866-025-03941-5
[25] [21] Ren J, Xia J, Zhang M, Liu C, Xu Y, Wu J, et al.. Automated identification of Salmonella serotype using MALDI-TOF mass spectrometry and machine learning techniques. Journal of Clinical Microbiology. 2025. DOI: https://doi.org/10.1128/jcm.00037-25
[26] [28] Normand A, Chaline A, Mohammad N, Godmer A, Acherar A, Huguenin A, et al.. Identification of a clonal population of Aspergillus flavus by MALDI-TOF mass spectrometry using deep learning. Scientific Reports. 2022. DOI: https://doi.org/10.1038/s41598-022-05647-4
[27] [38] Weis C, Cr J, K B. Machine learning for microbial identification and antimicrobial susceptibility testing on MALDI-TOF mass spectra: a systematic review.. Clinical Microbiology and Infection. 2020. DOI: https://doi.org/10.1016/j.cmi.2020.03.014
[28] [7] Khasapane N, Lekota K, Lehlohonolo M, Nkhebenyane J, Thekisoe O, Ramatla T. Antimicrobial resistance and virulence gene profiles of Enterococcus faecalis and Enterococcus faecium isolated from subclinical bovine mastitis milk and cow dung. Scientific Reports. 2025. DOI: https://doi.org/10.1038/s41598-025-32569-8
[29] [13] Pereira LBR, Garcia BLN, Fidelis CE, Barbosa KdS, Dantas STA, Rall VLM, et al.. Comparative Analysis of MALDI-TOF MS and RAPD for grouping Streptococcus agalactiae isolated from Subclinical Mastitis Isolates.. Microbial Pathogenesis. 2025. DOI: https://doi.org/10.1016/j.micpath.2025.107538
[30] [8] Tartor Y, Enany M, Elsheshtawy H, Kishk RM, Ali EM, Mahboub H, et al.. Dissemination of vancomycin-resistant Enterococcus faecalis and Enterococcus faecium between humans and fishes. Scientific Reports. 2026. DOI: https://doi.org/10.1038/s41598-026-36572-5
[31] [15] Pławińska-Czarnak J, Wódz K, Strzałkowska Z, Żychska M, Nowak T, Kwieciński A, et al.. Comparison of automatic methods MALDI-TOF, VITEK2 and manual methods for the identification of intestinal microbial communities on the example of samples from alpacas (Vicugna pacos). Journal of Veterinary Research. 2023. DOI: https://doi.org/10.2478/jvetres-2023-0051
[32] [16] Moreno L, Matajira CEC, Poor A, Mesquita RE, Gomes V, Silva AP, et al.. Identification through MALDI-TOF mass spectrometry and antimicrobial susceptibility profiling of bacterial pathogens isolated from sow urinary tract infection. Veterinary Quarterly. 2017. DOI: https://doi.org/10.1080/01652176.2017.1397302
[33] [29] Tyurin YA, Fassakhov R, Grigorieva T, Mustafin IG. [MICROBIAL COMPOSITION OF VARIOUS SURFACES OF SKIN DURING DEVELOP- MENT OF ATOPIC DERMATITIS BASED ON DATA FROM MALDI-TOF MASS-SPECTROMETRY IDENTIFICATION METHOD].. Zhurnal mikrobiologii, epidemiologii, i immunobiologii. 2016. DOI: https://doi.org/10.36233/0372-9311-2016-2-30-36
[34] [1] Liu E, Huang Y, Zhao Z, Teng T, Zhou H, Liu C, et al.. 2-Hydroxyfluorene triggers a vicious cycle of oxidative stress and gut dysbiosis, leading to intestinal injury in the polychaete Perinereis aibuhitensis. Comparative Biochemistry and Physiology Part C: Toxicology & Pharmacology. 2026. DOI: https://doi.org/10.1016/j.cbpc.2026.110556
[35] [4] Morgado DS, Costa GLd, Costa-Neto S, Souza IVd, Moratelli R, Oliveira MME. Polyphasic identification (MALDI-TOF + ITS) of mucosal yeasts in hybrid marmosets from Rio de Janeiro. Scientific Reports. 2026. DOI: https://doi.org/10.1038/s41598-026-43653-y
[36] [2] Hassen B. Antibiotic resistance in aquaculture: Contributions and perspectives of genomics. Aquaculture and Fisheries. 2026. DOI: https://doi.org/10.1016/j.aaf.2026.01.007
[37] [6] Bowers O, Killion HJ, Kuhlman K, Benjamin K, Studyvin J, Sondgeroth KS. Antimicrobial resistance phenotypes of E. coli from livestock and wildlife in Wyoming, U.S.A. BMC Veterinary Research. 2026. DOI: https://doi.org/10.1186/s12917-026-05386-y
[38] [5] Kizerwetter-Świda M, Garbowska M, Stefańska I, Pławińska-Czarnak J. False positive results obtained with latex agglutination identification of Staphylococcus aureus caused by Macrococcus caseolyticus and Mammaliicoccus fleurettii. Scientific Reports. 2025. DOI: https://doi.org/10.1038/s41598-025-31847-9
[39] [9] Adukkadukkam S, Suppala CS, Palpandi K, Kumar PA, Lingappasamy DT, Mariappa M, et al.. Antimicrobial Resistance in Acinetobacter baumannii Isolated From Healthy Animals. Acta Pathologica, Microbiologica et Immunologica Scandinavica (APMIS). 2026. DOI: https://doi.org/10.1111/apm.70186
[40] [22] Niyomdecha N, Sungvaraporn S, Pinmuang A, Mungkornkaew N, Saita T, Rodraksa W, et al.. Identification of bacteria on Thai banknotes and coins using MALDI-TOF mass spectrometry and their phenotypic antimicrobial susceptibility profiles. PeerJ. 2025. DOI: https://doi.org/10.7717/peerj.19465
[41] [32] Antunes TPB, Kemiyondo H. Artificial intelligence in veterinary diagnostic microbiology: Applications, opportunities and challenges in the one health era. International Health Sciences Review. 2026. DOI: https://doi.org/10.70164/ihsr.v2i2.146
[42] [43] Cortazzo V, Boza MdCP, Assanti VTG, Foglietta G, Vrenna G, Agosta M, et al.. The First Case of the Identification of a Microorganism Directly from Whole Blood Using MALDI-TOF Mass Spectrometry in an Onco-Hematological Pediatric Patient with Bloodstream Infection. Antibiotics. 2025. DOI: https://doi.org/10.3390/antibiotics14020149
[43] [44] Kato JM, Oliveira LMSd, Tanaka T, Santana L, Araújo EDMPDd, Yamamoto JH, et al.. Diagnostic value of paediatric blood culture bottle allied to MALDI-TOF mass spectrometry in the aetiological assessment of microbial keratitis.. Diagnostic microbiology and infectious disease. 2026. DOI: https://doi.org/10.1016/j.diagmicrobio.2026.117353
[44] [42] Sipahi N, Çevik Y. Differences and fingerprints of ESBL-producing E. coli from chicken faeces. Scientific Reports. 2025. DOI: https://doi.org/10.1038/s41598-025-14114-9
[45] [45] Montazeri S, Sharifinia F, Majd A, Ghasempour A, Akhavan A. Evaluation of Antibiotic Resistance in Mycobacterium tuberculosis Clinical Strains by Culture-Based Antibiogram, Target Gene Sequencing, and Matrix-Assisted Laser Desorption Ionization Time of Flight Mass Spectrometry (MALDI-TOF MS) Assay. Crescent Journal of Medical and Biological Sciences. 2025. DOI: https://doi.org/10.34172/cjmb.2025.30
[46] [46] Vitale RG, Hoog Sd, Stock F, Scudder CJ, Shahegh S, Aneke CI, et al.. Spectrum of clinically significant melanized fungi in NIH hospitalized patients and their antifungal susceptibility profiles.. Medical Mycology. 2025. DOI: https://doi.org/10.1093/mmy/myaf072
[47] [48] Sy I, Conrad L, Becker S. Recent Advances and Potential Future Applications of MALDI-TOF Mass Spectrometry for Identification of Helminths. Diagnostics. 2022. DOI: https://doi.org/10.3390/diagnostics12123035
[48] [47] Beker S, Demirbilek SK. Optimizing detection methods for MRSA isolated from mastitis cases and assessing virulence genes.. Research in Veterinary Science. 2025. DOI: https://doi.org/10.1016/j.rvsc.2025.105609
[49] [50] Han J, Tang Y, Wu S, Chen Y, Zou Z, Zeng H, et al.. Identification of Virulence Genes and Antibiotic Resistance in Extraintestinal Pathogenic Escherichia coli Isolated from Broiler Carcasses Using MALDI-TOF MS. Pathogens. 2025. DOI: https://doi.org/10.3390/pathogens14050501
[50] [49] Lauková A, Ščerbová J, Focková V, Stojanov I, Simonová MP, Prodanov-Radulović J. Assessing Virulence Factor Genes in Pig-Derived Escherichia coli from the Region of Vojvodina Treated with Postbiotic Substance and Herbal Essential Oils. Pathogens. 2026. DOI: https://doi.org/10.3390/pathogens15020215
[51] [51] Ludwiczak A, Sibińska E, Adamczyk I, Wasicki M, Pryshchepa O, Złoch M, et al.. Integration of MALDI‐TOF MS and 16S rRNA Analysis for Identification of Plant‐Based Fermentation‐Associated Microbiota. Environmental Microbiology Reports. 2026. DOI: https://doi.org/10.1111/1758-2229.70237
[52] [52] Cojkic A, Hansson I, Morrell J. Bacterial survival below zero: Impact of storage time on bacterial viability in bull semen.. Cryobiology. 2025. DOI: https://doi.org/10.1016/j.cryobiol.2025.105233
[53] [54] Ludwiczak A, Zieliński T, Sibińska E, Czeszewska-Rosiak G, Złoch M, Rudnicka J, et al.. Comparative analysis of microbial contamination in diesel fuels using MALDI-TOF MS. Scientific Reports. 2025. DOI: https://doi.org/10.1038/s41598-025-87713-1
[54] [53] Orak F, Karakaya E, Satıcıoğlu IB, Akar M, Güran C, Abay S, et al.. Anaerobic bacteria from bloodstream infections: Identification and antibacterial susceptibility testing in a single center in Türkiye.. Acta Microbiologica et Immunologica Hungarica. 2025. DOI: https://doi.org/10.1556/030.2025.02476
[55] [55] Błońska D, Buszewski B. Characterization of Honey Microbiome Using MALDI-TOF Mass Spectrometry and Physicochemical Study. Molecules. 2025. DOI: https://doi.org/10.3390/molecules30061266
[56] [57] Cao Q, Deng Z, Li M, Zhu S, Huo Y, Dong H, et al.. Integrated metagenomic and metabolomic analyses reveal tenacissoside G as a potential non-antimicrobial treatment for bovine endometritis. Microbiome. 2025. DOI: https://doi.org/10.1186/s40168-025-02264-x
[57] [56] Onder B, Erdogdu B, Onder S, Ozbek T. Developing a proteomic approach through structural characterization of bacteriophage K. Bioorganic Chemistry. 2026. DOI: https://doi.org/10.1016/j.bioorg.2026.109858 *** Disclaimer: This article is for educational and informational purposes only. It is not intended to substitute for professional veterinary advice, diagnosis, treatment, or regulatory guidance. Always consult a licensed veterinarian or qualified specialist regarding animal health, disease diagnosis, and therapeutic decisions.